

Real-world outcomes from a series of patients with late onset Pompe disease who switched from alglucosidase alfa to avalglucosidase alfa
- PMID: 38313679
- PMCID: PMC10834735
- DOI: 10.3389/fgene.2024.1309146
Real-world outcomes from a series of patients with late onset Pompe disease who switched from alglucosidase alfa to avalglucosidase alfa
Abstract
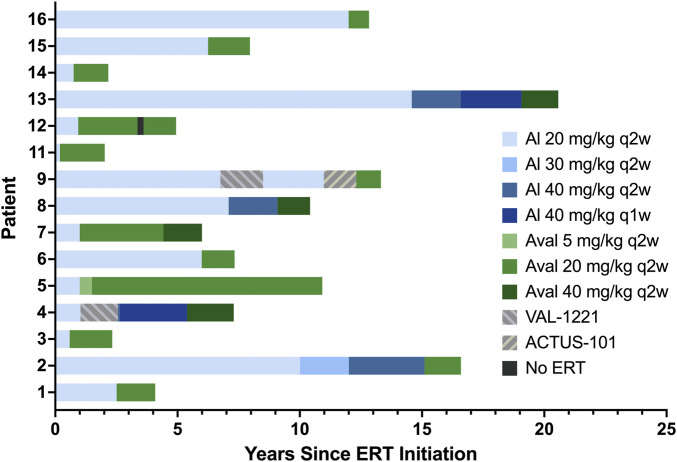
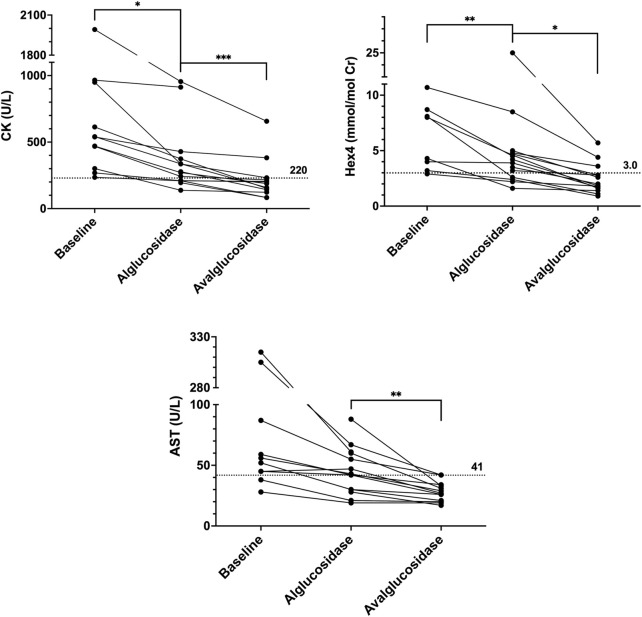
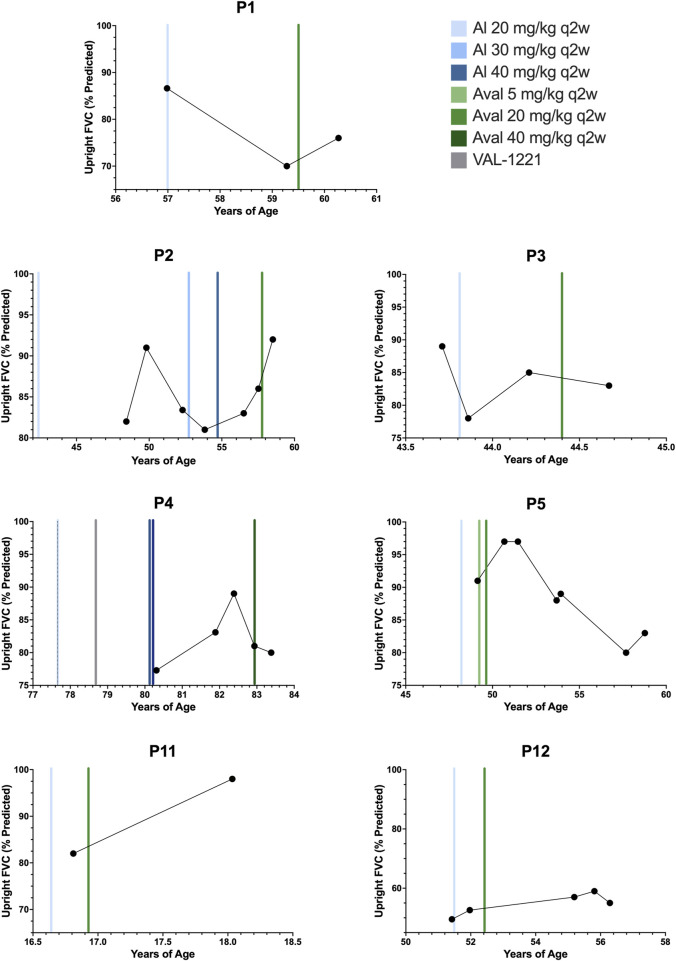
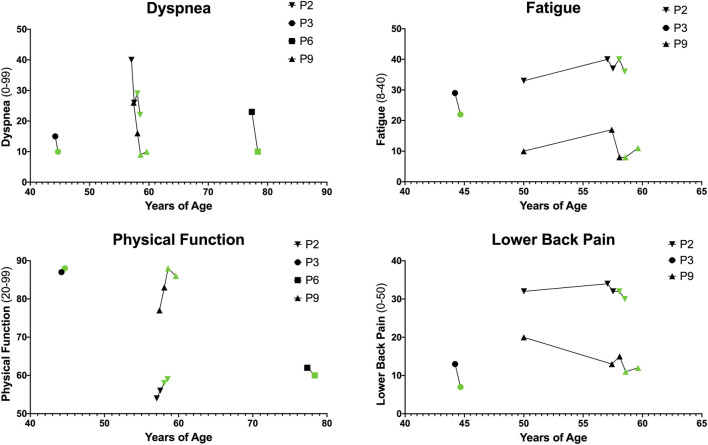
Introduction: Pompe disease is an inherited, progressive neuromuscular disorder caused by deficiency of lysosomal acid α-glucosidase and accumulation of glycogen in tissues, resulting in cellular dysfunction, muscle damage, and functional disabilities. Enzyme replacement therapy with alglucosidase alfa (Myozyme/Lumizyme) has led to better outcomes, but many patients have plateaued or declined despite treatment. The second-generation ERT avalglucosidase alfa (Nexviazyme) was designed to have enhanced cellular uptake via the conjugation of additional bis-mannose-6-phosphate residues. There have been trials comparing the efficacy of alglucosidase and avalglucosidase, but there remains a need for more real-world data on patients who switched from alglucosidase to avalglucosidase. Methods: A chart review was conducted on n = 15 patients with late-onset Pompe disease followed at a single center who switched from alglucosidase to avalglucosidase and continued for at least 6 months. Results: A total of n = 8/15 patients received alglucosidase for more than 3 years prior to switching, and n = 7/15 received it for more than 5 years prior to switching. There were statistically significant improvements in CK, Hex4, and AST with mean differences of -104.8 U/L, -3.0 mmol/molCr, and -14.7 U/L, respectively, post-switch. 6-Minute Walk Test; comfortable gait speed; Gait, Stairs, Gower, Chair; and Quick Motor Function Test scores improved or stabilized in most patients post-switch (n = 8/12, n = 11/12, n = 9/12, n =7/11, respectively). Of n = 7 patients with pulmonary function testing, n = 4/7 had improved upright FVC. Patient-reported outcomes revealed improvements in dyspnea (n = 4/4), physical function (n = 3/4), fatigue (n = 2/3), and lower back pain (n = 3/3). Avalglucosidase was well tolerated without infusion-associated reactions, and all n = 7 patients on home infusions continued receiving ERT at home. Anti-drug antibodies were seen in n = 9/10 of patients on alglucosidase and n = 8/13 of those on avalglucosidase, with titers below 12,800 in a majority of patients. We also present the first outcome data for a patient with LOPD who is non-ambulatory and a full-time wheelchair user; she demonstrated meaningful improvements in quality of life and motor function with the switch. Discussion: In summary, improved outcomes were seen in most patients, with a subset whose decline persisted. This study presents evidence that switching from alglucosidase to avalglucosidase may be associated with improved outcomes in certain patients with LOPD.
Keywords: alglucosidase alfa; avalglucosidase alfa; enzyme replacement therapy; glycogen storage disease type 2; late-onset Pompe disease (LOPD); lysosomal storage disease (LSD).
Copyright © 2024 Carter, Boggs, Case and Kishnani.
Conflict of interest statement
PK has received research/grant support from Sanofi Genzyme and Amicus Therapeutics. PK has received consulting fees and honoraria from Sanofi Genzyme, Amicus Therapeutics, Maze Therapeutics, Bayer and Asklepios Biopharmaceutical, Inc. (AskBio). PK is a member of the Pompe and Gaucher Disease Registry Advisory Board for Sanofi Genzyme, Pompe Disease Advisory Board for Amicus Therapeutics, and Advisory Board for Baebies. PK has equity with Maze Therapeutics and has held equity in Asklepios Biopharmaceuticals, and may receive milestone payments related to that equity in the future. TB has participated in research funded by Sanofi Genzyme, Amicus Therapeutics and Asklepios Biopharmaceutical, Inc. (AskBio). TB is a member of the Pompe Physical Therapy Advisory Board for Sanofi Genzyme, and 2021 Pompe Disease Advisory Board for Sanofi Genzyme. LC has received honoraria from Genzyme Corporation of Sanofi, Pfizer, and Sarepta for presentations given; has participated in research funded by Amicus, AskBio, AveXis, Biogen, Genzyme Corporation of Sanofi, NS Pharma, Pfizer, PTC Therapeutics, Reveragen, and TRiNDS (Therapeutic Research in Neuromuscular Disorders Solutions); and is a member of the North American Pompe Registry Board of Advisors. The remaining author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
References
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous