

PI3K/mTOR is a therapeutically targetable genetic dependency in diffuse intrinsic pontine glioma
- PMID: 38319732
- PMCID: PMC10940093
- DOI: 10.1172/JCI170329
PI3K/mTOR is a therapeutically targetable genetic dependency in diffuse intrinsic pontine glioma
Abstract
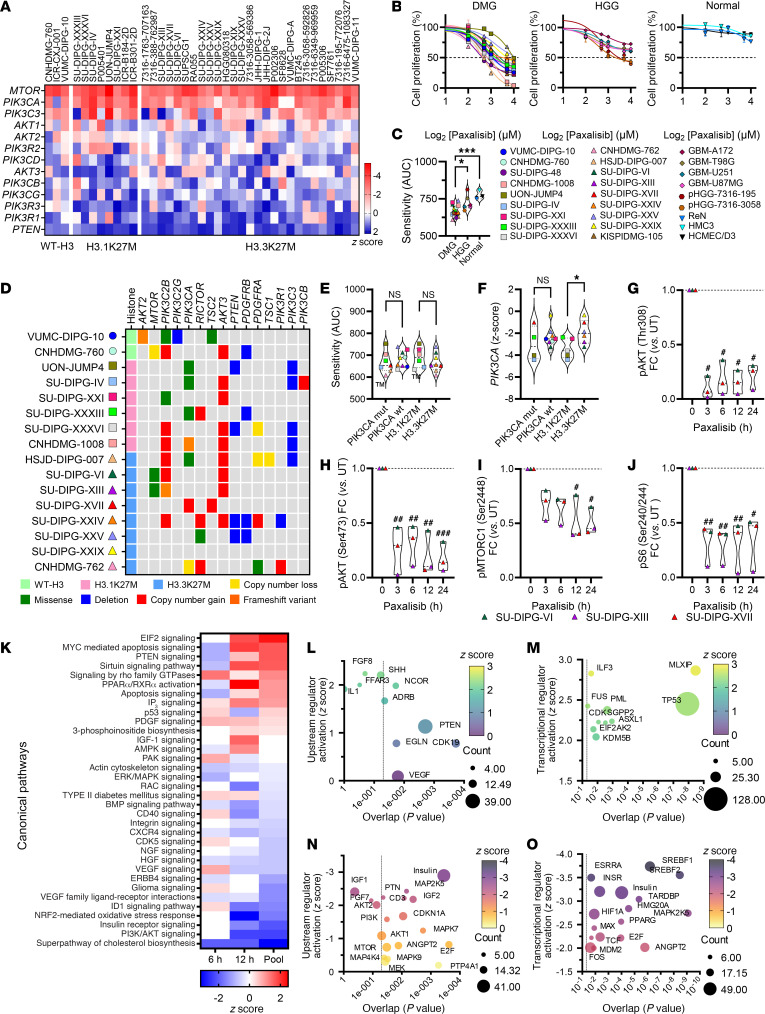
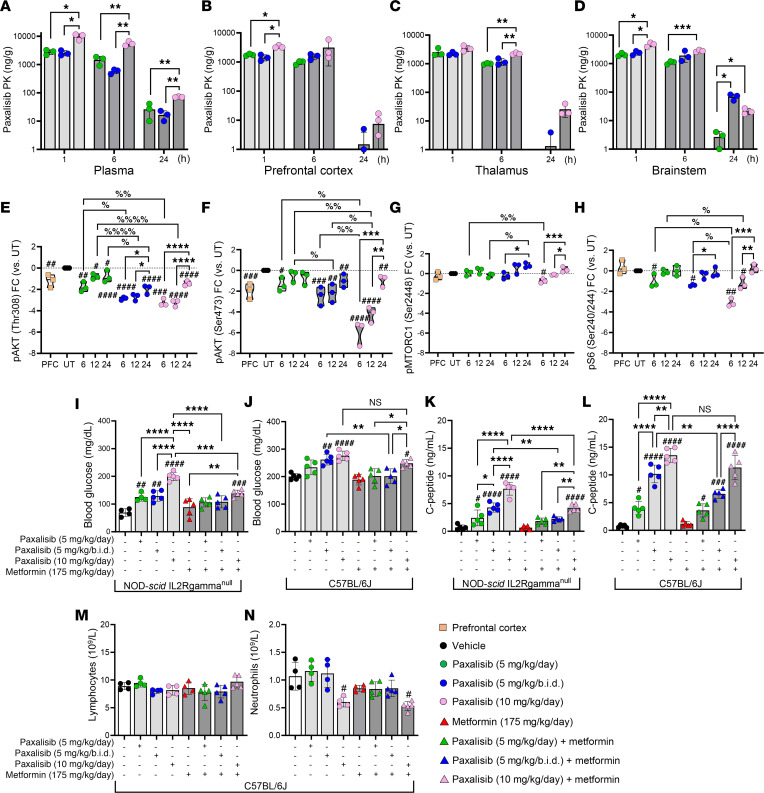
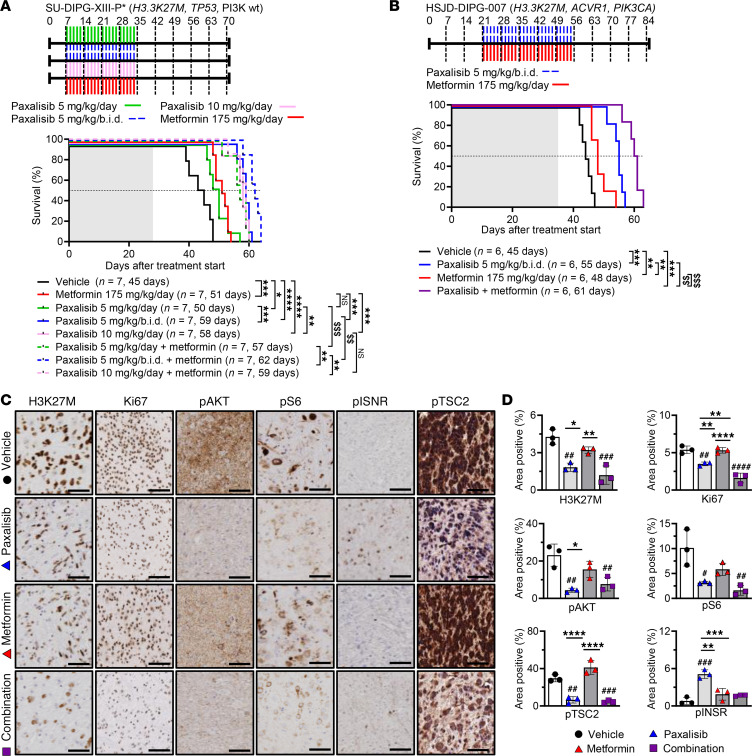
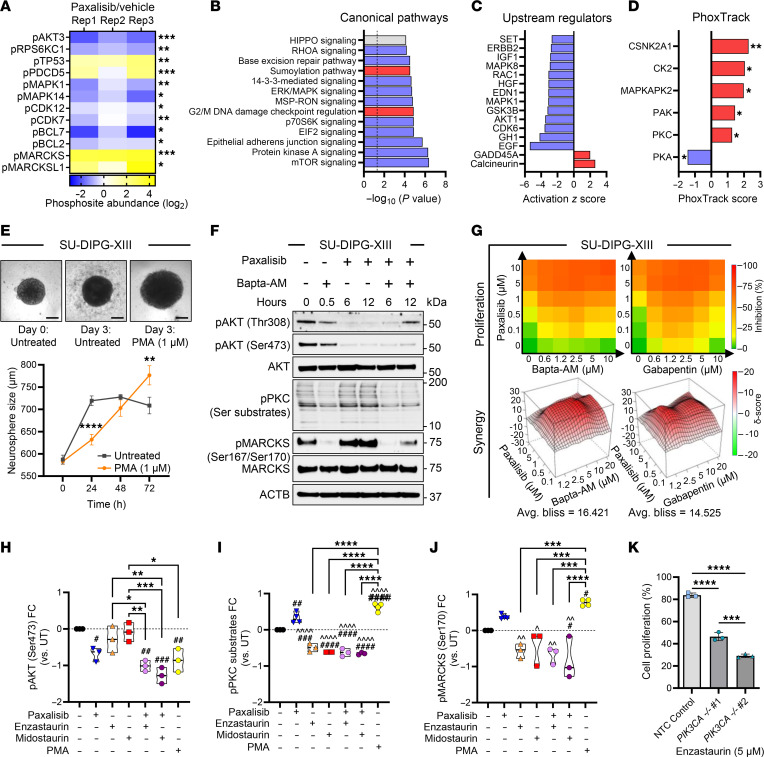
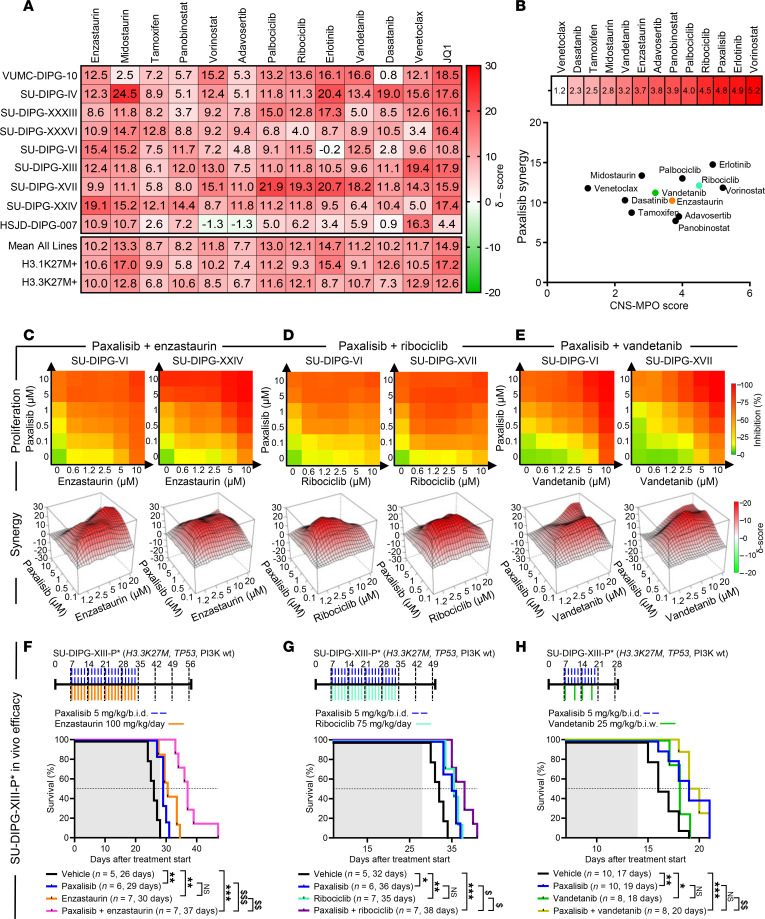
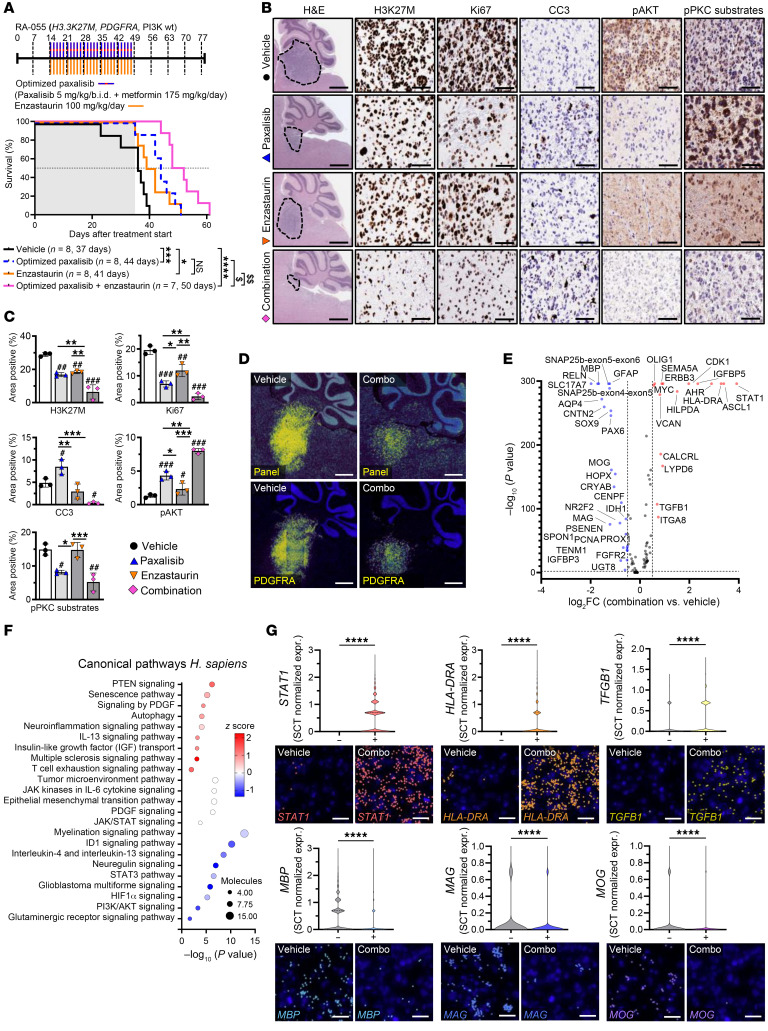
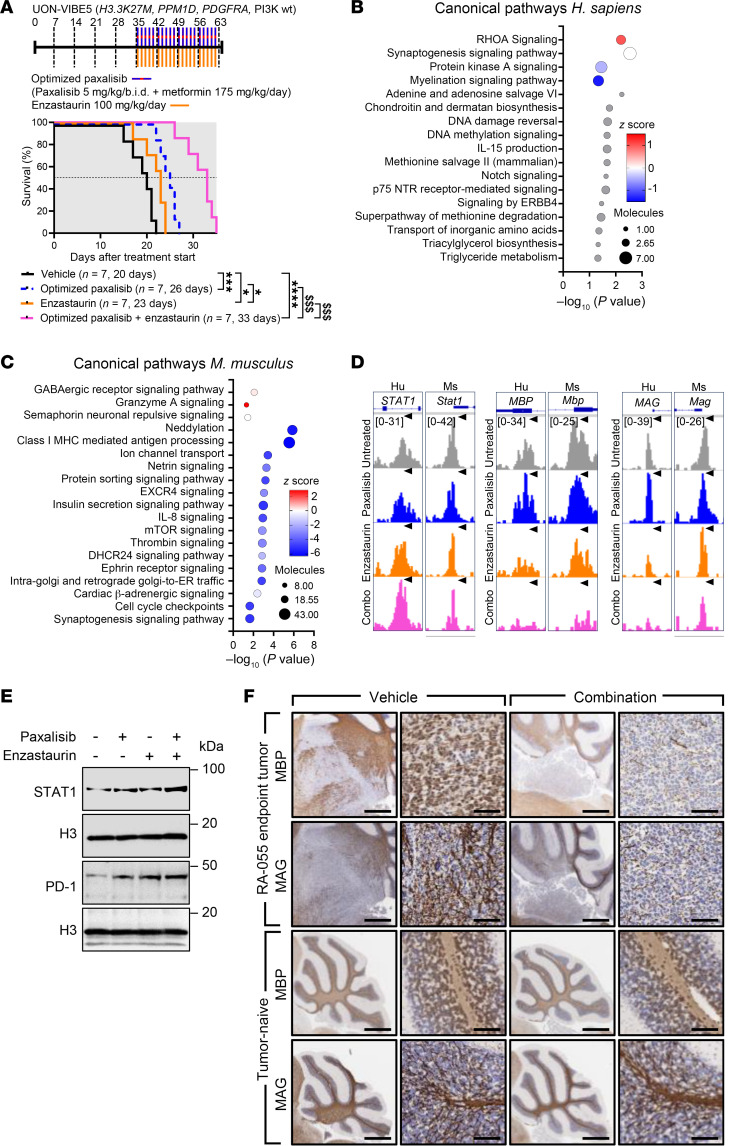
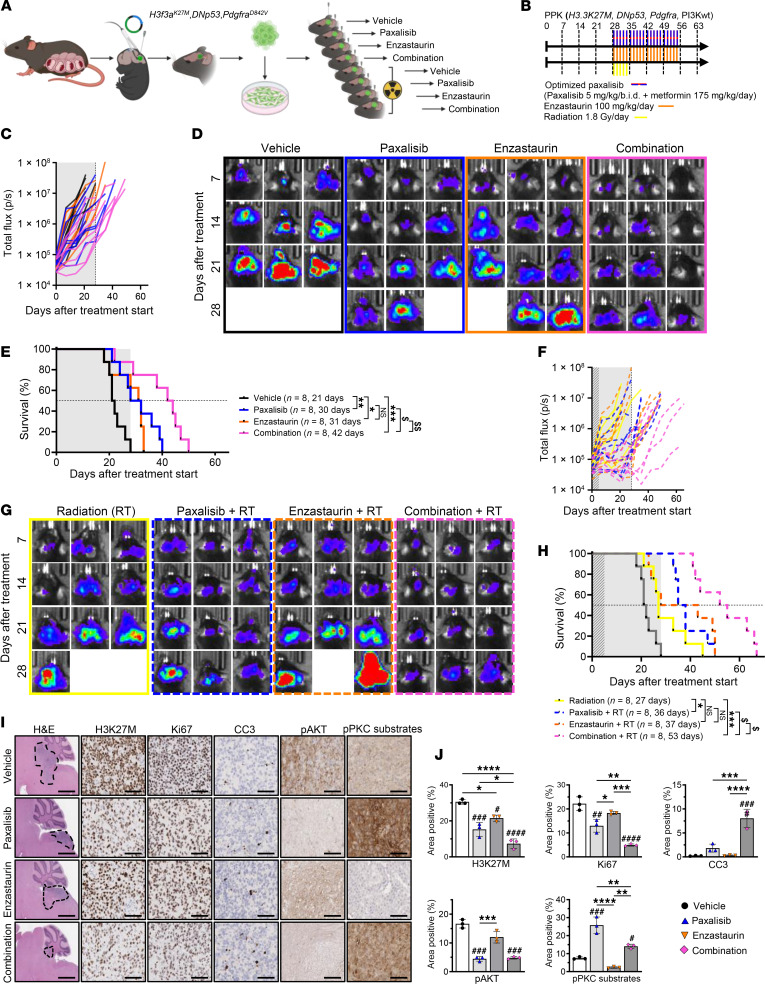
Diffuse midline glioma (DMG), including tumors diagnosed in the brainstem (diffuse intrinsic pontine glioma; DIPG), are uniformly fatal brain tumors that lack effective treatment. Analysis of CRISPR/Cas9 loss-of-function gene deletion screens identified PIK3CA and MTOR as targetable molecular dependencies across patient derived models of DIPG, highlighting the therapeutic potential of the blood-brain barrier-penetrant PI3K/Akt/mTOR inhibitor, paxalisib. At the human-equivalent maximum tolerated dose, mice treated with paxalisib experienced systemic glucose feedback and increased insulin levels commensurate with patients using PI3K inhibitors. To exploit genetic dependence and overcome resistance while maintaining compliance and therapeutic benefit, we combined paxalisib with the antihyperglycemic drug metformin. Metformin restored glucose homeostasis and decreased phosphorylation of the insulin receptor in vivo, a common mechanism of PI3K-inhibitor resistance, extending survival of orthotopic models. DIPG models treated with paxalisib increased calcium-activated PKC signaling. The brain penetrant PKC inhibitor enzastaurin, in combination with paxalisib, synergistically extended the survival of multiple orthotopic patient-derived and immunocompetent syngeneic allograft models; benefits potentiated in combination with metformin and standard-of-care radiotherapy. Therapeutic adaptation was assessed using spatial transcriptomics and ATAC-Seq, identifying changes in myelination and tumor immune microenvironment crosstalk. Collectively, this study has identified what we believe to be a clinically relevant DIPG therapeutic combinational strategy.
Keywords: Brain cancer; Drug therapy; Oncogenes; Oncology; Therapeutics.
Figures
Comment in
- Just a spoonful of metformin helps the medicine go down
References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
