

Transcriptome-wide association study of the plasma proteome reveals cis and trans regulatory mechanisms underlying complex traits
- PMID: 38320554
- PMCID: PMC10940016
- DOI: 10.1016/j.ajhg.2024.01.006
Transcriptome-wide association study of the plasma proteome reveals cis and trans regulatory mechanisms underlying complex traits
Abstract
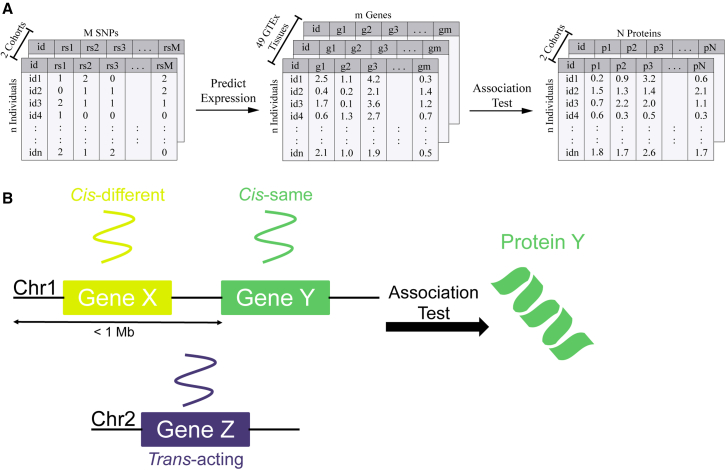
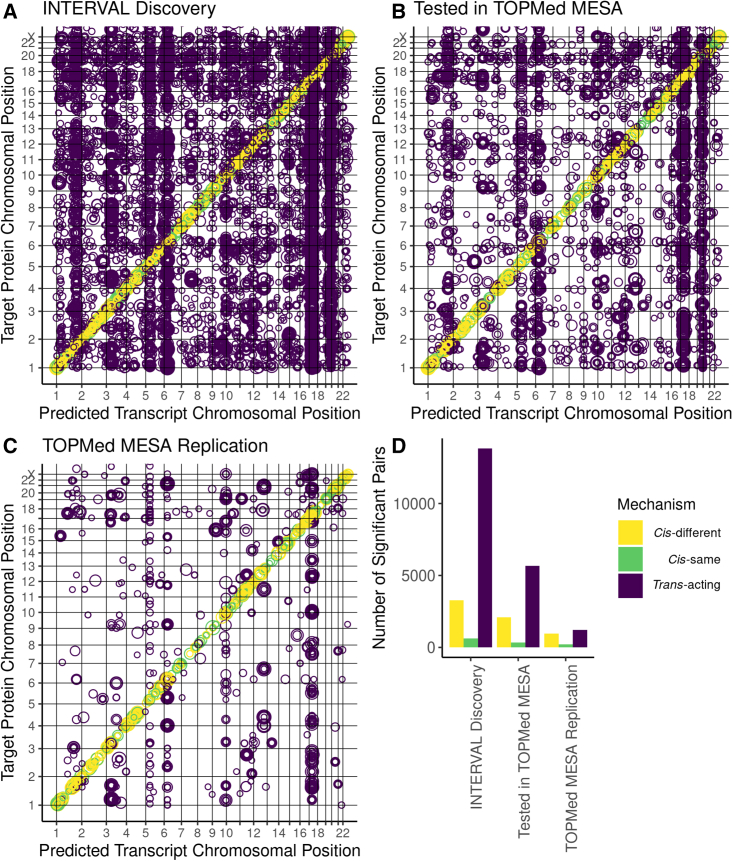
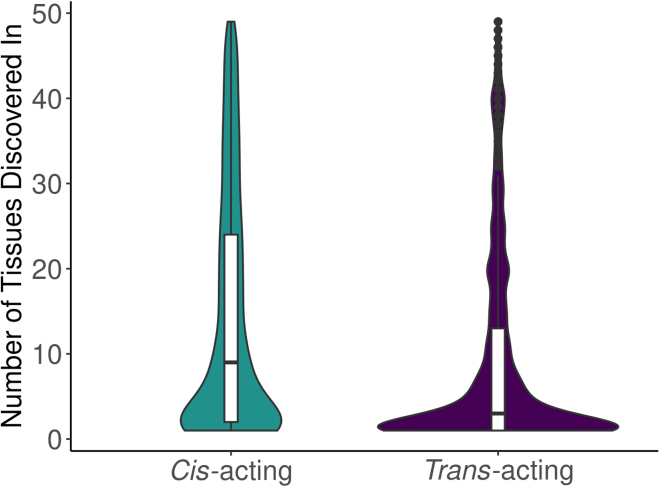
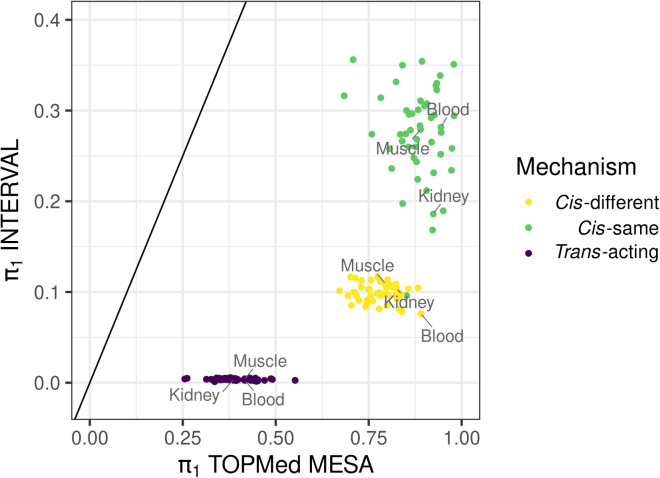
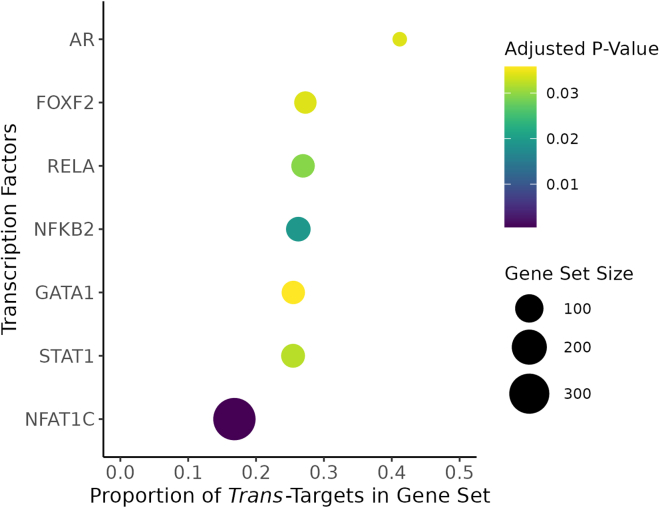
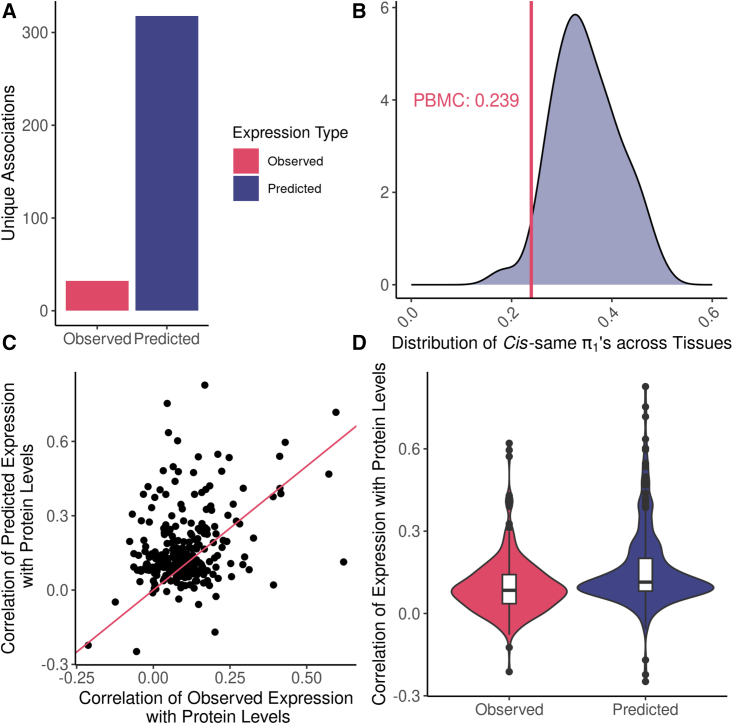
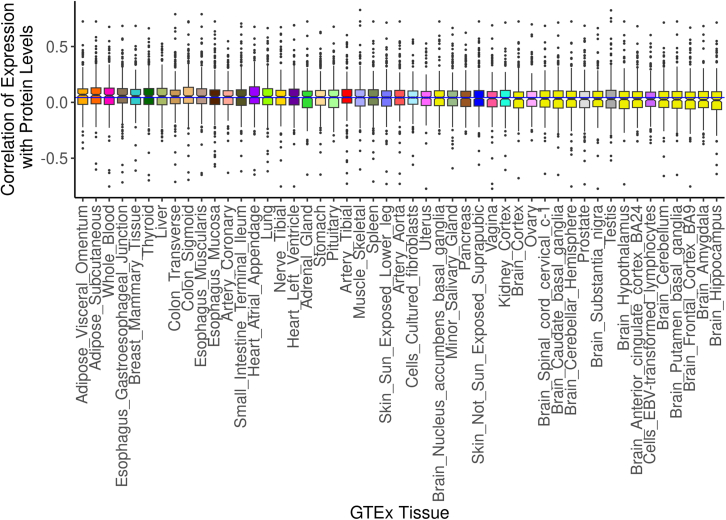
Regulation of transcription and translation are mechanisms through which genetic variants affect complex traits. Expression quantitative trait locus (eQTL) studies have been more successful at identifying cis-eQTL (within 1 Mb of the transcription start site) than trans-eQTL. Here, we tested the cis component of gene expression for association with observed plasma protein levels to identify cis- and trans-acting genes that regulate protein levels. We used transcriptome prediction models from 49 Genotype-Tissue Expression (GTEx) Project tissues to predict the cis component of gene expression and tested the predicted expression of every gene in every tissue for association with the observed abundance of 3,622 plasma proteins measured in 3,301 individuals from the INTERVAL study. We tested significant results for replication in 971 individuals from the Trans-omics for Precision Medicine (TOPMed) Multi-Ethnic Study of Atherosclerosis (MESA). We found 1,168 and 1,210 cis- and trans-acting associations that replicated in TOPMed (FDR < 0.05) with a median expected true positive rate (π1) across tissues of 0.806 and 0.390, respectively. The target proteins of trans-acting genes were enriched for transcription factor binding sites and autoimmune diseases in the GWAS catalog. Furthermore, we found a higher correlation between predicted expression and protein levels of the same underlying gene (R = 0.17) than observed expression (R = 0.10, p = 7.50 × 10-11). This indicates the cis-acting genetically regulated (heritable) component of gene expression is more consistent across tissues than total observed expression (genetics + environment) and is useful in uncovering the function of SNPs associated with complex traits.
Keywords: TWAS; autoimmune diseases; cis-eQTL; gene expression; genetic prediction; heritability; plasma proteome; trans-eQTL; transcription factors.
Copyright © 2024 American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests The authors declare no competing interests.
Figures
References
-
- Strunz T., Grassmann F., Gayán J., Nahkuri S., Souza-Costa D., Maugeais C., Fauser S., Nogoceke E., Weber B.H.F. A mega-analysis of expression quantitative trait loci (eQTL) provides insight into the regulatory architecture of gene expression variation in liver. Sci. Rep. 2018;8:5865. - PMC - PubMed
-
- Võsa U., Claringbould A., Westra H.-J., Bonder M.J., Deelen P., Zeng B., Kirsten H., Saha A., Kreuzhuber R., Yazar S., et al. Large-scale cis- and trans-eQTL analyses identify thousands of genetic loci and polygenic scores that regulate blood gene expression. Nat. Genet. 2021;53:1300–1310. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
