

Anti-aquaporin-4 immune complex stimulates complement-dependent Th17 cytokine release in neuromyelitis optica spectrum disorders
- PMID: 38326464
- PMCID: PMC10850367
- DOI: 10.1038/s41598-024-53661-5
Anti-aquaporin-4 immune complex stimulates complement-dependent Th17 cytokine release in neuromyelitis optica spectrum disorders
Abstract
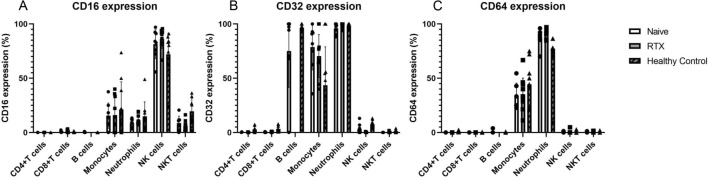
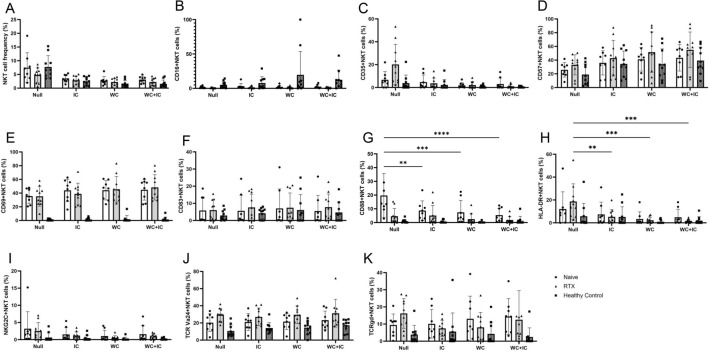
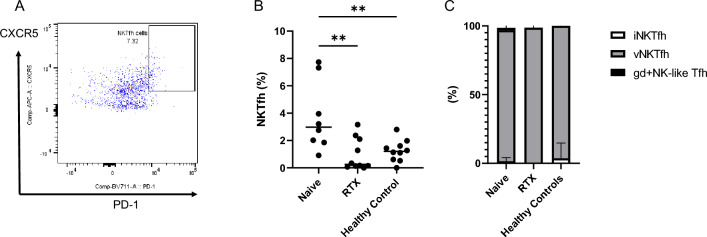
Proinflammatory cytokines, such as (IL: interleukin) IL-6 and IL-17A, and complement fixation are critical in the immunopathogenesis of neuromyelitis optica spectrum disorders (NMOSD). Blocking the IL-6 receptor or the C5 complement pathway reduces relapse risk. However, the role of interleukin (IL)-6 and complement in aquaporin-4 (AQP4) autoimmunity remains unclear. To investigate the role of the anti-AQP4 immunoglobulin (AQP4-IgG)/AQP4 immunocomplex on the induction and profile of ex vivo cytokine and surface marker expression in peripheral blood mononuclear cells (PBMC) culture. Isolated PBMCs obtained from 18 patients with AQP4-IgG-seropositive-NMOSD (8 treatment-naive, 10 rituximab-treated) or ten healthy controls were cultured with AQP4-immunocomplex with or without complement. Changes in PBMC surface markers and cytokine expression were profiled using flow cytometry and ELISA. PBMCs derived from treatment-naive NMOSD patients stimulated with a complex mixture of serum complement proteins produced significant elevations of IL-17A and IL-6. Rituximab-treated patients also exhibited higher IL-6 but not IL-17A release. IL-6 and IL-17A elevations are not observed without complement. Co-stimulation of PBMCs with AQP4-IgG/AQP4 immunocomplex and complement prompts a Th17-biased response consistent with the inflammatory paradigm observed in NMOSD. A possible inflammation model is proposed via antigen-specific autoreactive peripheral blood cells, including NK/NKT cells.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures




References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
