

IDHwt glioblastomas can be stratified by their transcriptional response to standard treatment, with implications for targeted therapy
- PMID: 38326875
- PMCID: PMC10848526
- DOI: 10.1186/s13059-024-03172-3
IDHwt glioblastomas can be stratified by their transcriptional response to standard treatment, with implications for targeted therapy
Abstract
Background: Glioblastoma (GBM) brain tumors lacking IDH1 mutations (IDHwt) have the worst prognosis of all brain neoplasms. Patients receive surgery and chemoradiotherapy but tumors almost always fatally recur.
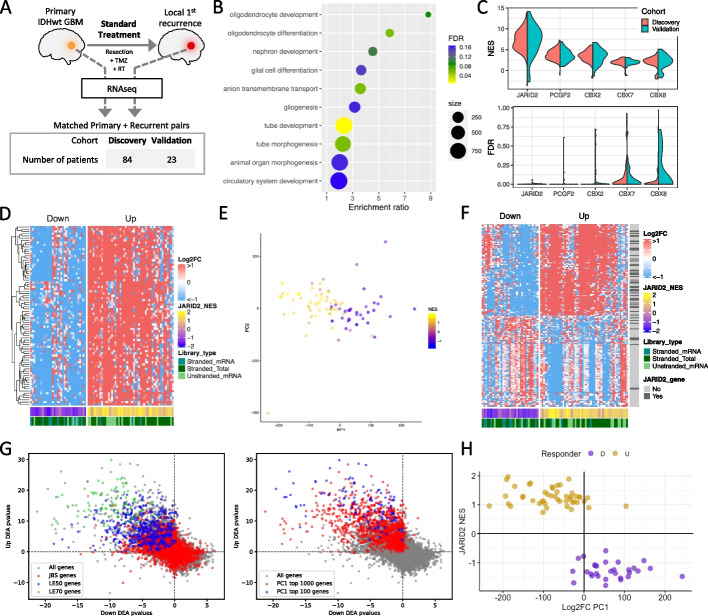
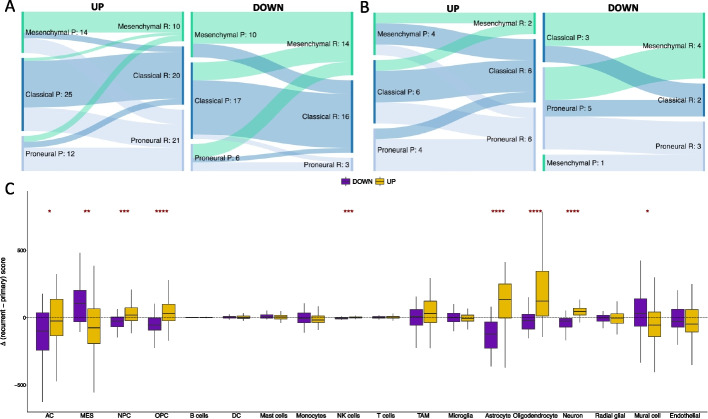
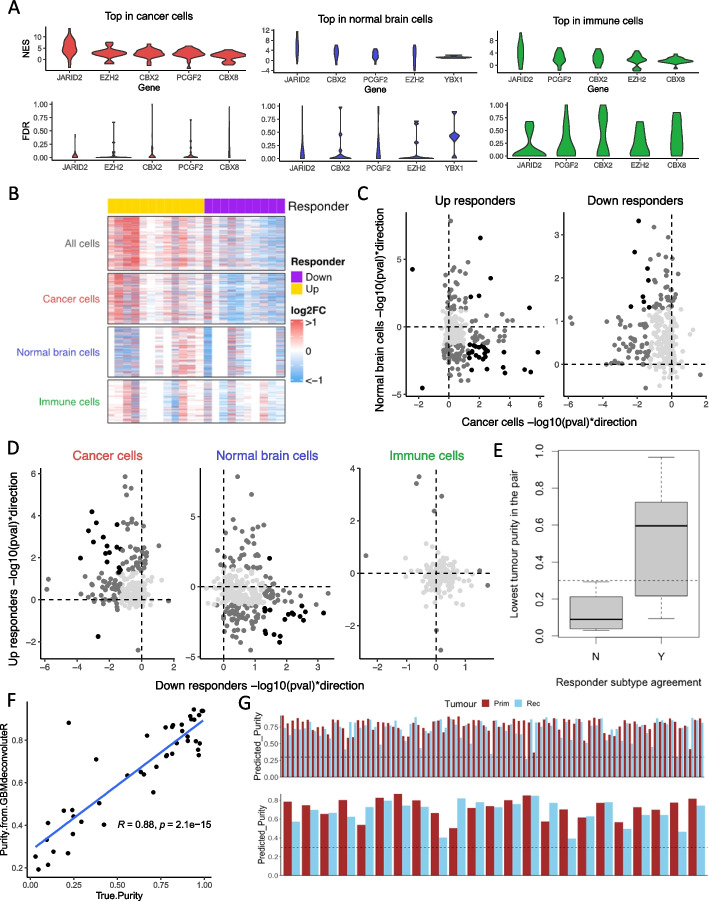
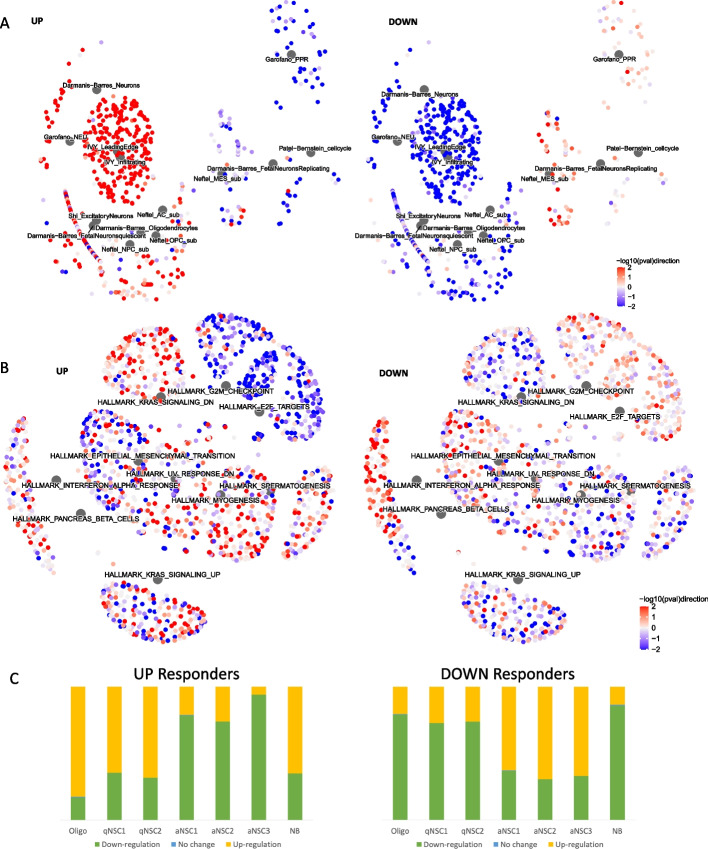
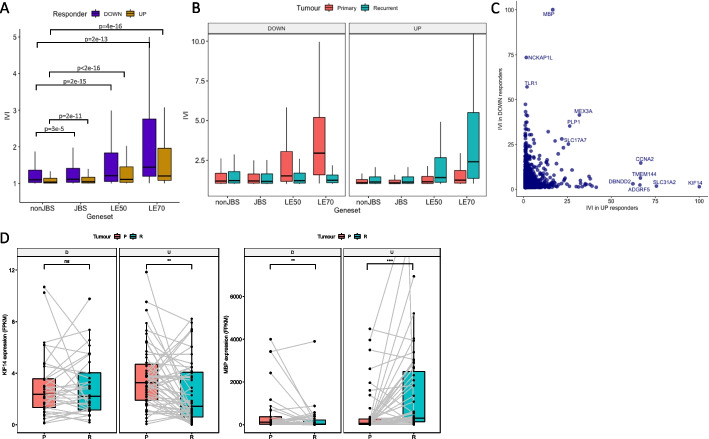
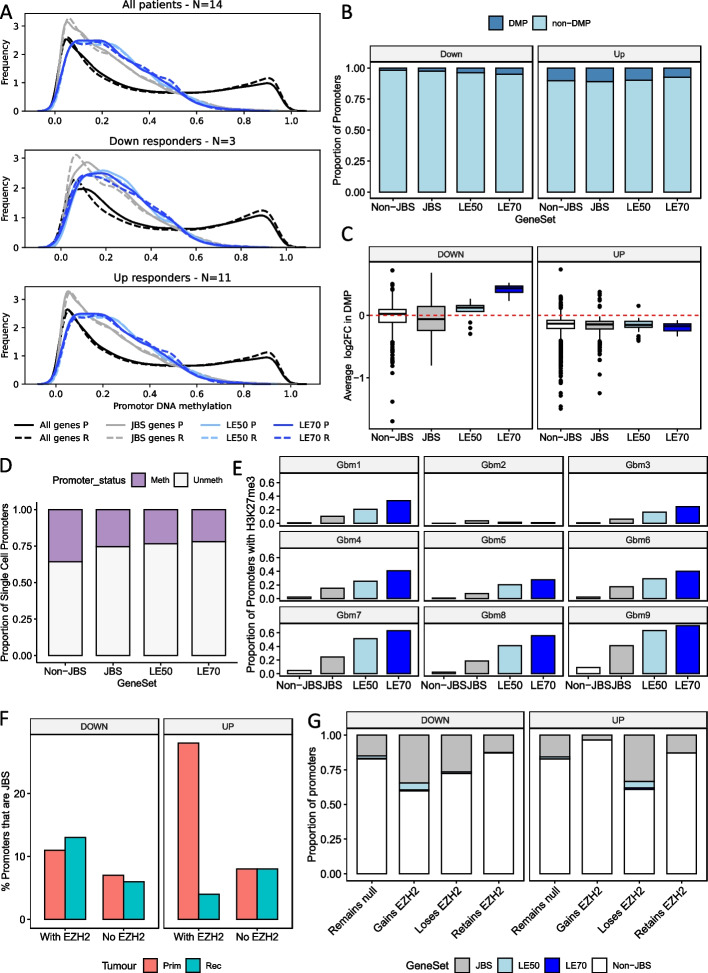
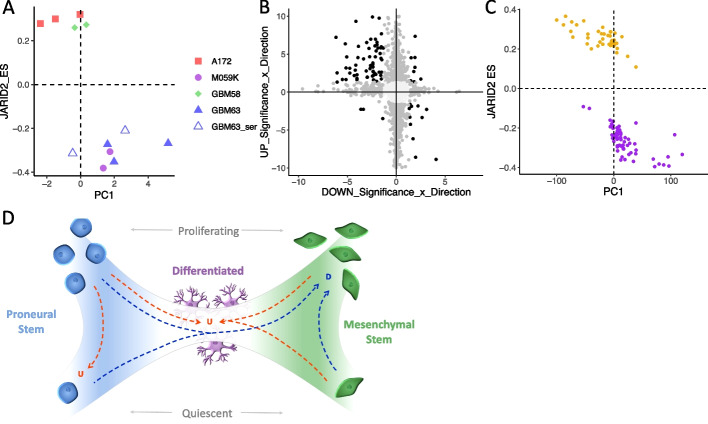
Results: Using RNA sequencing data from 107 pairs of pre- and post-standard treatment locally recurrent IDHwt GBM tumors, we identify two responder subtypes based on longitudinal changes in gene expression. In two thirds of patients, a specific subset of genes is upregulated from primary to recurrence (Up responders), and in one third, the same genes are downregulated (Down responders), specifically in neoplastic cells. Characterization of the responder subtypes indicates subtype-specific adaptive treatment resistance mechanisms that are associated with distinct changes in the tumor microenvironment. In Up responders, recurrent tumors are enriched in quiescent proneural GBM stem cells and differentiated neoplastic cells, with increased interaction with the surrounding normal brain and neurotransmitter signaling, whereas Down responders commonly undergo mesenchymal transition. ChIP-sequencing data from longitudinal GBM tumors suggests that the observed transcriptional reprogramming could be driven by Polycomb-based chromatin remodeling rather than DNA methylation.
Conclusions: We show that the responder subtype is cancer-cell intrinsic, recapitulated in in vitro GBM cell models, and influenced by the presence of the tumor microenvironment. Stratifying GBM tumors by responder subtype may lead to more effective treatment.
© 2024. The Author(s).
Conflict of interest statement
RGWV is a consultant for NeuroTrials, Inc, and Stellanova Therapeutics. AD is the director of AD Bioinformatics Ltd. LFS is on the Scientific Advisory Board for CoSyne Therapeutics Ltd.
Figures
References
-
- Korber V, Yang J, Barah P, Wu Y, Stichel D, Gu Z, Fletcher MNC, Jones D, Hentschel B, Lamszus K, et al. Evolutionary trajectories of IDH(WT) glioblastomas reveal a common path of early tumorigenesis instigated years ahead of initial diagnosis. Cancer Cell. 2019;35(692–704):e612. - PubMed
-
- Wang L, Babikir H, Muller S, Yagnik G, Shamardani K, Catalan F, Kohanbash G, Alvarado B, Di Lullo E, Kriegstein A, et al. The phenotypes of proliferating glioblastoma cells reside on a single axis of variation. Cancer Discov. 2019;9:1708–1719. doi: 10.1158/2159-8290.CD-19-0329. - DOI - PMC - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
