

p17/C18-ceramide-mediated mitophagy is an endogenous neuroprotective response in preclinical and clinical brain injury
- PMID: 38328780
- PMCID: PMC10847724
- DOI: 10.1093/pnasnexus/pgae018
p17/C18-ceramide-mediated mitophagy is an endogenous neuroprotective response in preclinical and clinical brain injury
Abstract
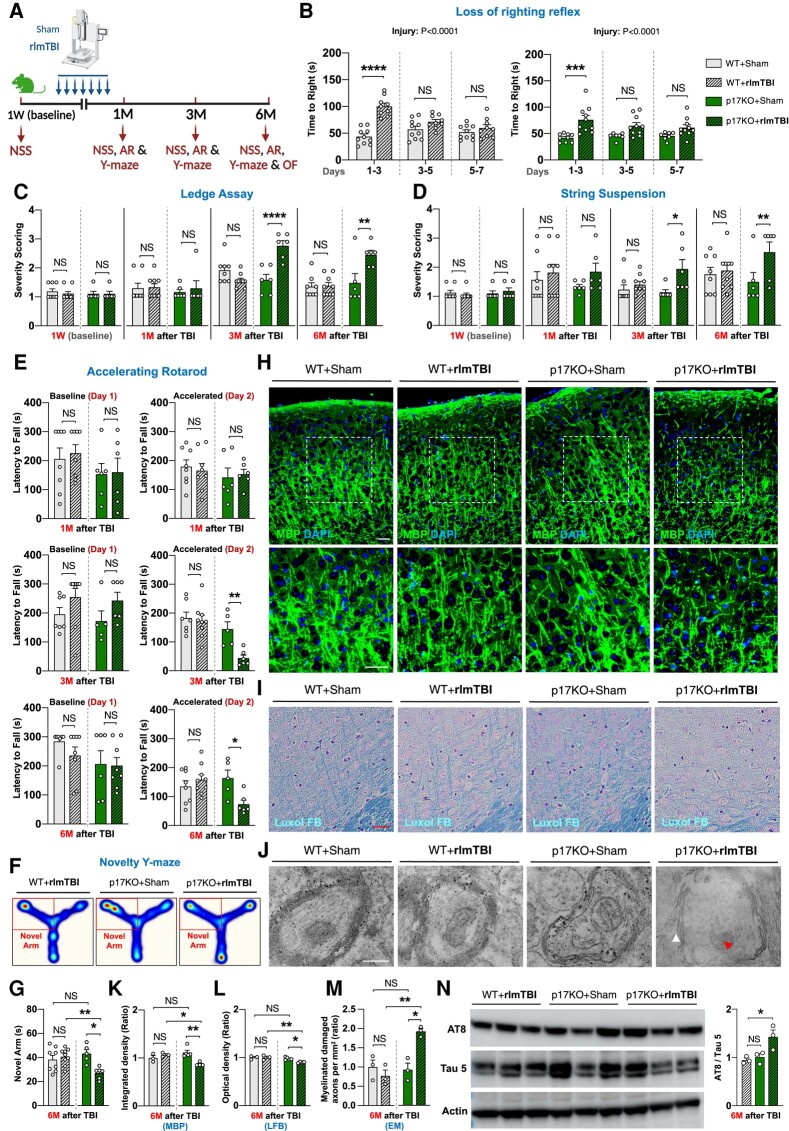
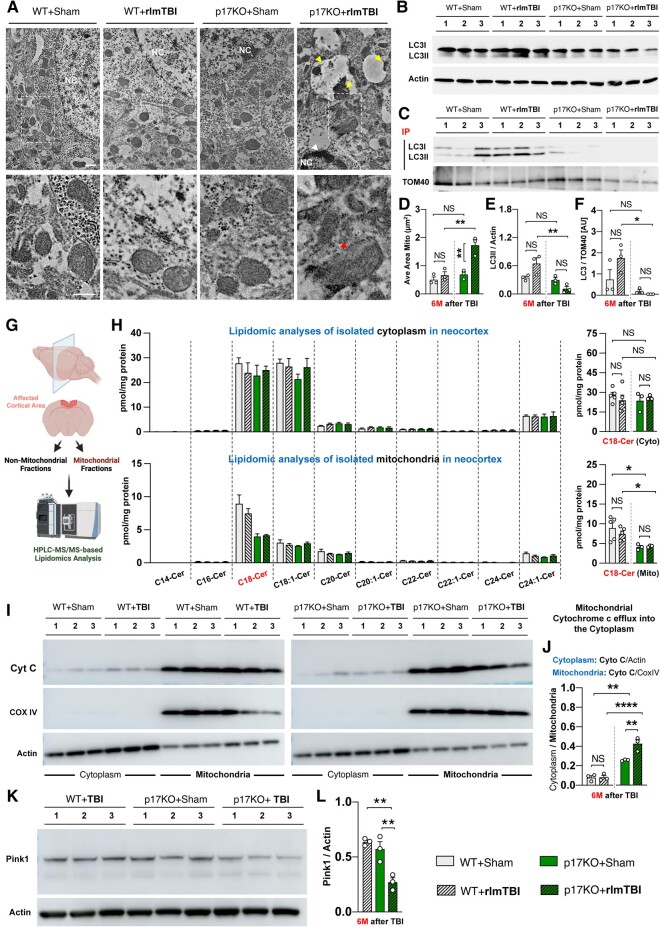
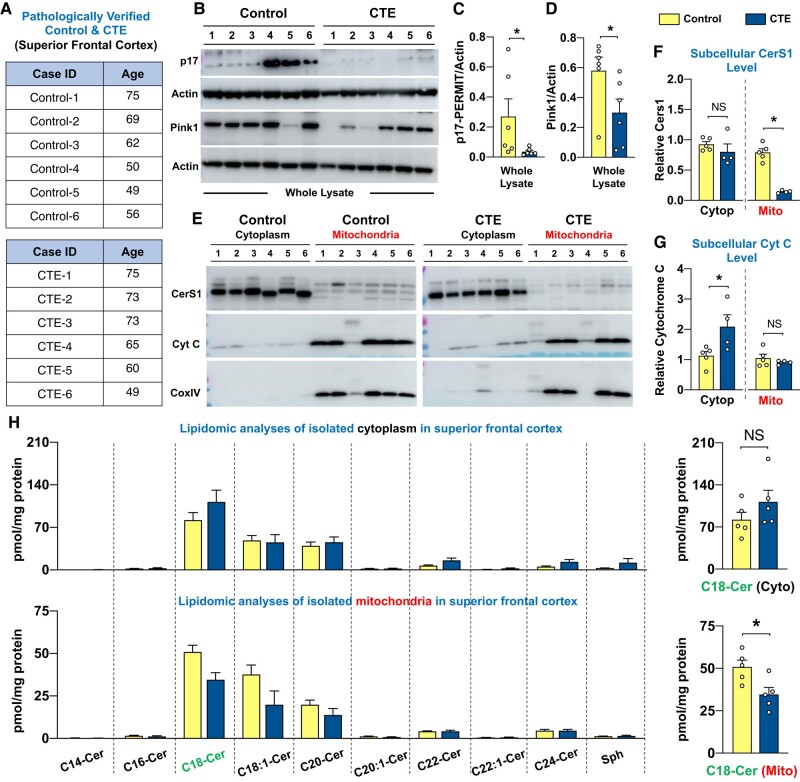
Repeat concussions (or repetitive mild traumatic brain injury [rmTBI]) are complex pathological processes consisting of a primary insult and long-term secondary complications and are also a prerequisite for chronic traumatic encephalopathy (CTE). Recent evidence implies a significant role of autophagy-mediated dysfunctional mitochondrial clearance, mitophagy, in the cascade of secondary deleterious events resulting from TBI. C18-ceramide, a bioactive sphingolipid produced in response to cell stress and damage, and its synthesizing enzyme (CerS1) are precursors to selective stress-mediated mitophagy. A transporter, p17, mediates the trafficking of CerS1, induces C18-ceramide synthesis in the mitochondrial membrane, and acts as an elimination signal in cell survival. Whether p17-mediated mitophagy occurs in the brain and plays a causal role in mitochondrial quality control in secondary disease development after rmTBI are unknown. Using a novel repetitive less-than-mild TBI (rlmTBI) injury paradigm, ablation of mitochondrial p17/C18-ceramide trafficking in p17 knockout (KO) mice results in a loss of C18-ceramide-induced mitophagy, which contributes to susceptibility and recovery from long-term secondary complications associated with rlmTBI. Using a ceramide analog with lipid-selenium conjugate drug, LCL768 restored mitophagy and reduced long-term secondary complications, improving cognitive deficits in rlmTBI-induced p17KO mice. We obtained a significant reduction of p17 expression and a considerable decrease of CerS1 and C18-ceramide levels in cortical mitochondria of CTE human brains compared with age-matched control brains. These data demonstrated that p17/C18-ceramide trafficking is an endogenous neuroprotective mitochondrial stress response following rlmTBI, thus suggesting a novel prospective strategy to interrupt the CTE consequences of concussive TBI.
© The Author(s) 2024. Published by Oxford University Press on behalf of National Academy of Sciences.
Figures



References
-
- Lye TC, Shores EA. 2000. Traumatic brain injury as a risk factor for Alzheimer's disease: a review. Neuropsychol Rev. 10(2):115–129. - PubMed
-
- Nordstrom P, Michaelsson K, Gustafson Y, Nordstrom A. 2014. Traumatic brain injury and young onset dementia: a nationwide cohort study. Ann Neurol. 75(3):374–381. - PubMed
-
- Coronado VG, et al. 2011. Surveillance for traumatic brain injury-related deaths–United States, 1997–2007. MMWR Surveill Summ. 60(5):1–32. - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
