

Human Fc gamma receptor IIIA blockade inhibits platelet destruction in a humanized murine model of ITP
- PMID: 38330193
- PMCID: PMC11007428
- DOI: 10.1182/bloodadvances.2023012155
Human Fc gamma receptor IIIA blockade inhibits platelet destruction in a humanized murine model of ITP
Abstract
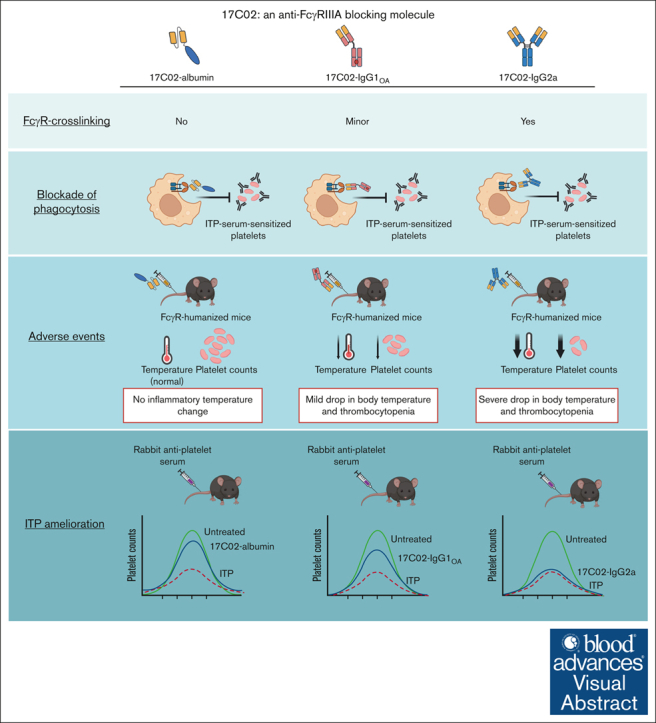
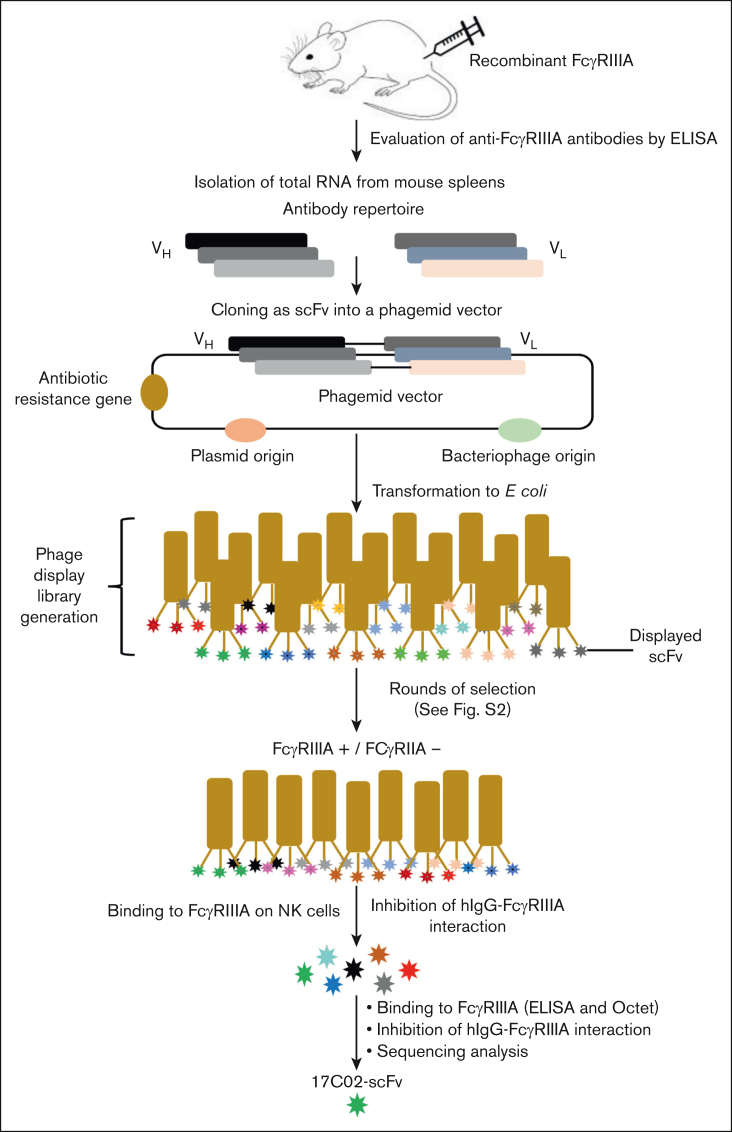
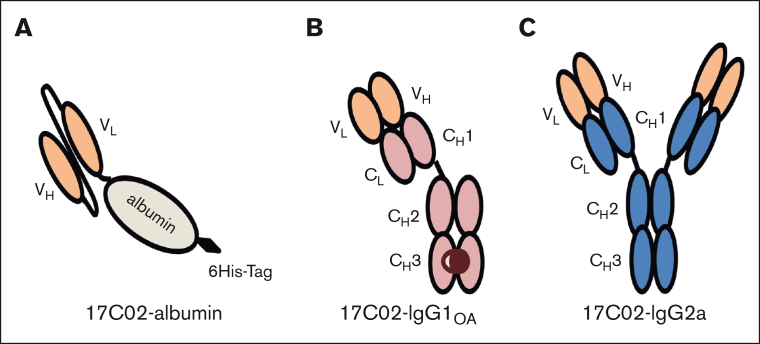
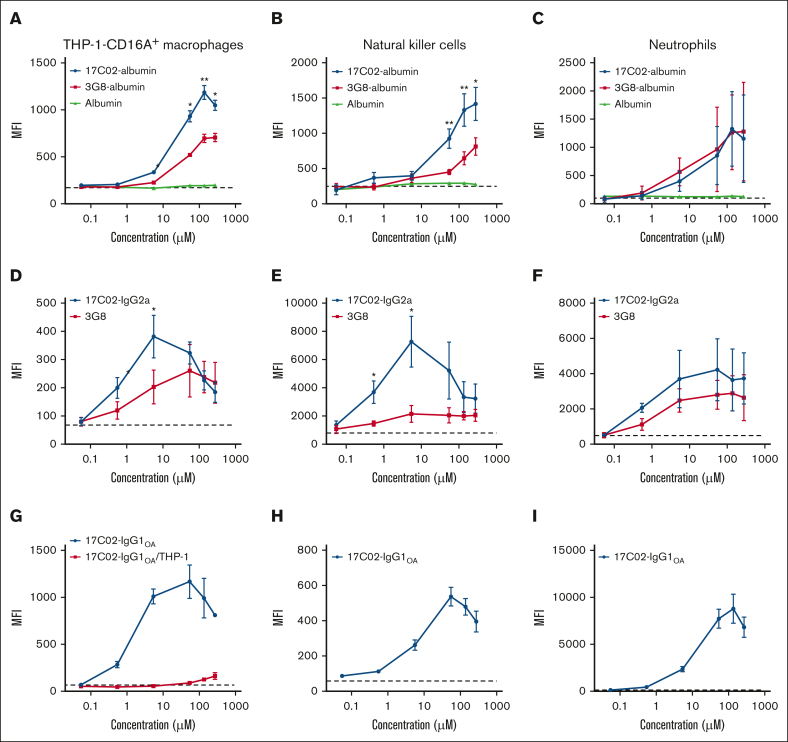
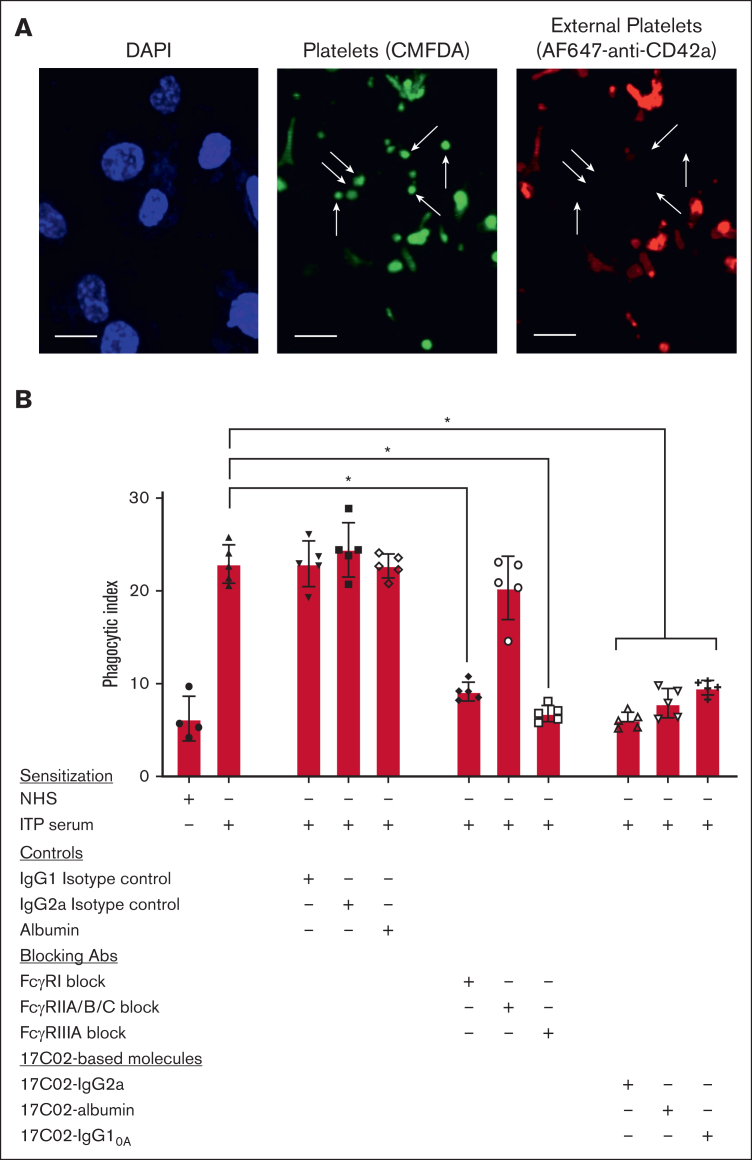
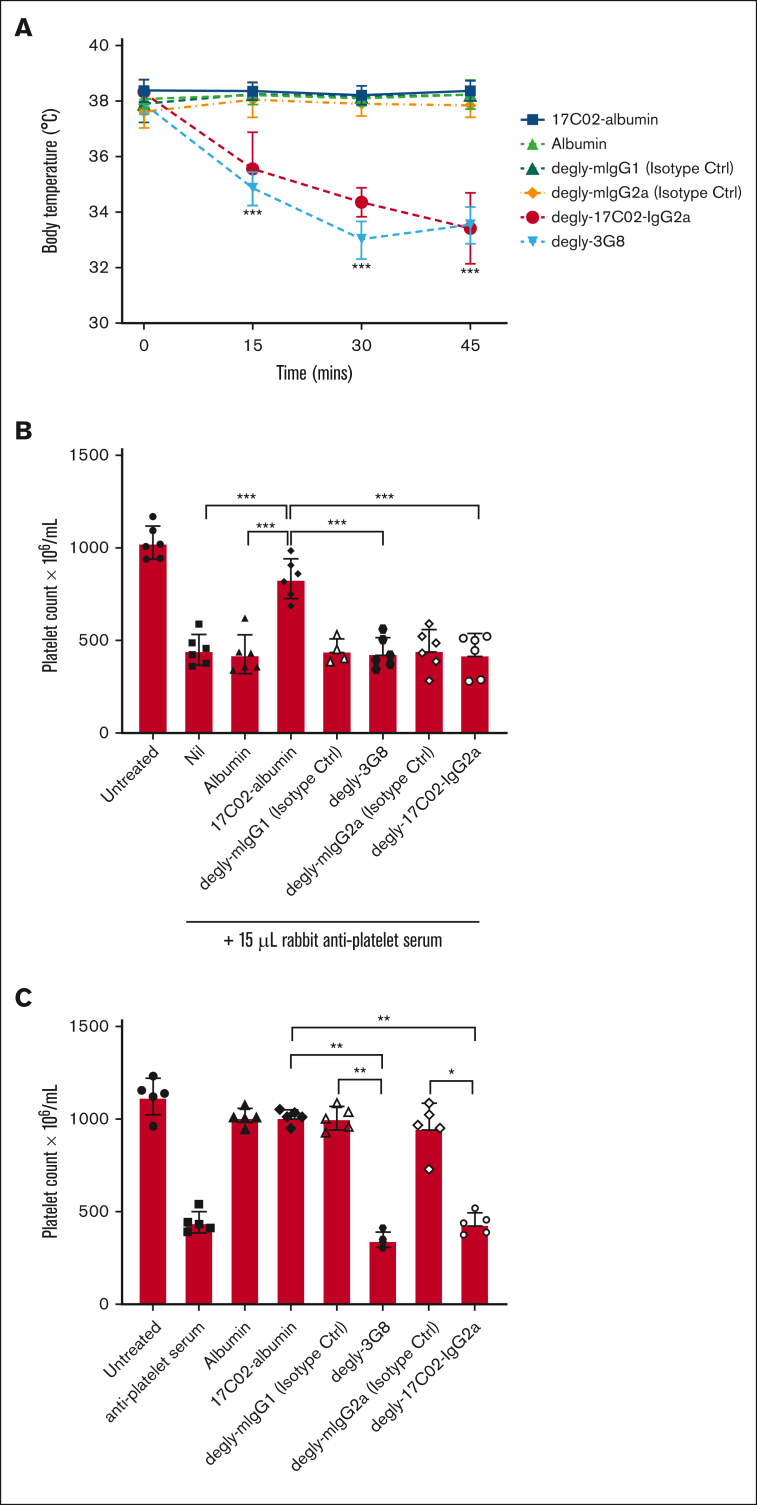
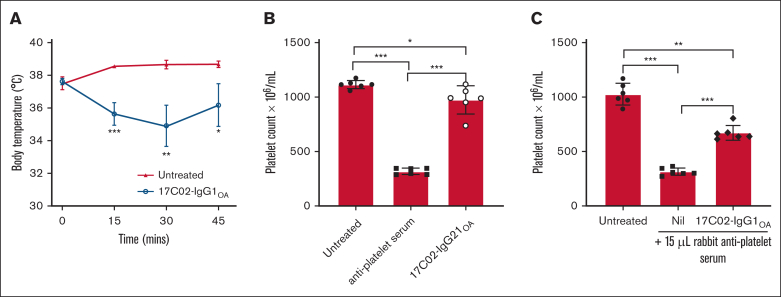
Fc gamma receptor (FcγR) IIIA is an important receptor for immunoglobulin G (IgG) and is involved in immune defense mechanisms as well as tissue destruction in some autoimmune diseases including immune thrombocytopenia (ITP). FcγRIIIA on macrophages can trigger phagocytosis of IgG-sensitized platelets, and prior pilot studies observed blockade of FcγRIIIA increased platelet counts in patients with ITP. Unfortunately, although blockade of FcγRIIIA in patients with ITP increased platelet counts, its engagement by the blocking antibody drove serious adverse inflammatory reactions. These adverse events were postulated to originate from the antibody's Fc and/or bivalent nature. The blockade of human FcγRIIIA in vivo with a monovalent construct lacking an active Fc region has not yet been achieved. To effectively block FcγRIIIA in vivo, we developed a high affinity monovalent single-chain variable fragment (scFv) that can bind and block human FcγRIIIA. This scFv (17C02) was expressed in 3 formats: a monovalent fusion protein with albumin, a 1-armed human IgG1 antibody, and a standard bivalent mouse (IgG2a) antibody. Both monovalent formats were effective in preventing phagocytosis of ITP serum-sensitized human platelets. In vivo studies using FcγR-humanized mice demonstrated that both monovalent therapeutics were also able to increase platelet counts. The monovalent albumin fusion protein did not have adverse event activity as assessed by changes in body temperature, whereas the 1-armed antibody induced some changes in body temperature even though the Fc region function was impaired by the Leu234Ala and Leu235Ala mutations. These data demonstrate that monovalent blockade of human FcγRIIIA in vivo can potentially be a therapeutic strategy for patients with ITP.
© 2024 by The American Society of Hematology. Licensed under Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International (CC BY-NC-ND 4.0), permitting only noncommercial, nonderivative use with attribution. All other rights reserved.
Conflict of interest statement
Conflict-of-interest disclosure: Patent applications describing the 17C02-based molecules are assigned to the Canadian Blood Services with the participation of adMare BioInnovations and Unity Health/St. Michael’s Hospital. A.H.L. has had research funding from CSL and has other patents on IV immunoglobulin alternatives. The remaining authors declare no competing financial interests.
Figures
References
-
- Semple JW, Rebetz J, Maouia A, Kapur R. An update on the pathophysiology of immune thrombocytopenia. Curr Opin Hematol. 2020;27(6):423–429. - PubMed
-
- Kurlander RJ, Ellison DM, Hall J. The blockade of Fc receptor-mediated clearance of immune complexes in vivo by a monoclonal antibody (2.4G2) directed against Fc receptors on murine leukocytes. J Immunol. 1984;133(2):855–862. - PubMed
-
- Clarkson SB, Bussel JB, Kimberly RP, Valinsky JE, Nachman RL, Unkeless JC. Treatment of refractory immune thrombocytopenic purpura with an anti-Fc gamma-receptor antibody. N Engl J Med. 1986;314(19):1236–1239. - PubMed
