

Highly Accurate Prediction of NMR Chemical Shifts from Low-Level Quantum Mechanics Calculations Using Machine Learning
- PMID: 38331423
- PMCID: PMC11702896
- DOI: 10.1021/acs.jctc.3c01256
Highly Accurate Prediction of NMR Chemical Shifts from Low-Level Quantum Mechanics Calculations Using Machine Learning
Abstract
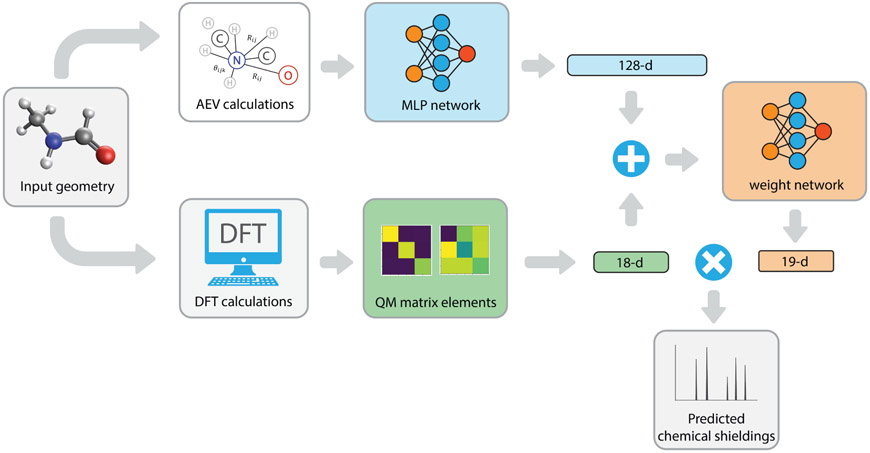
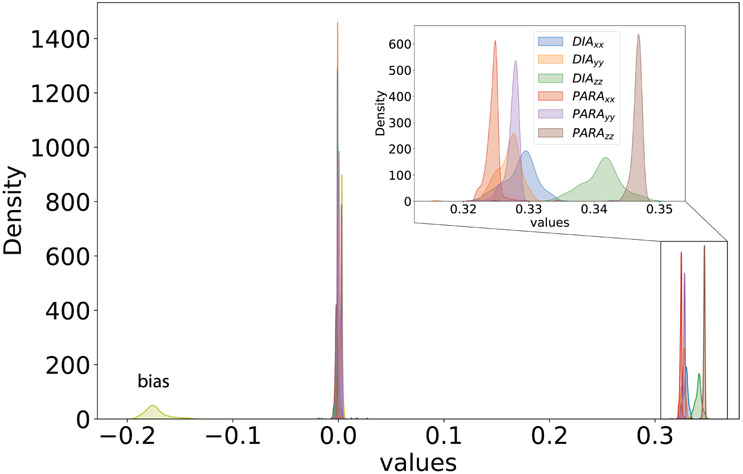
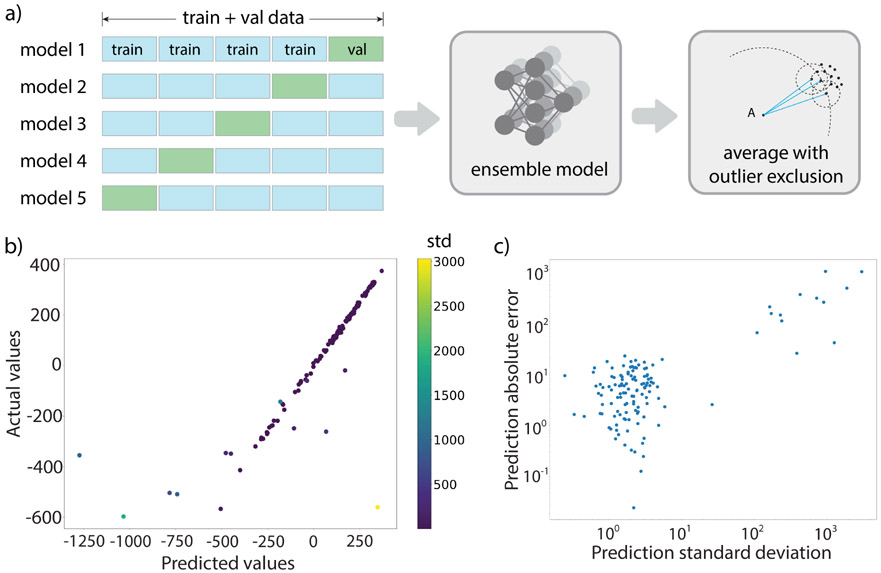
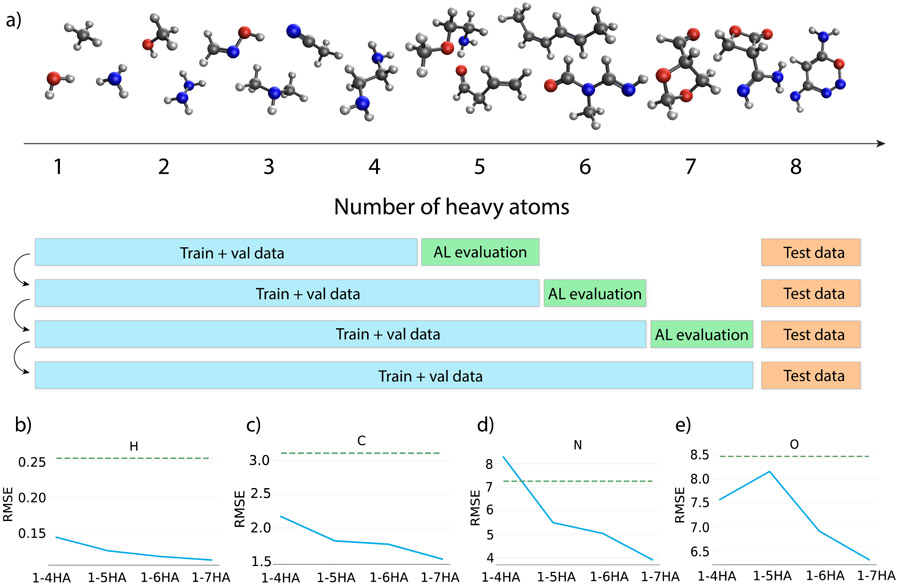
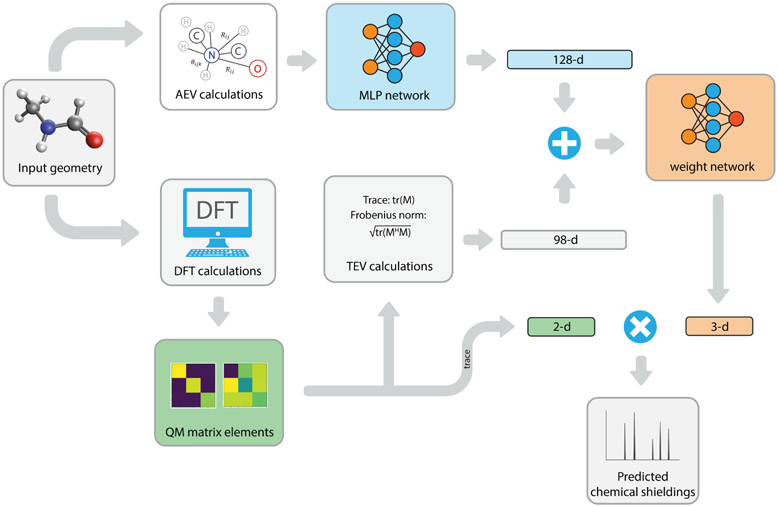
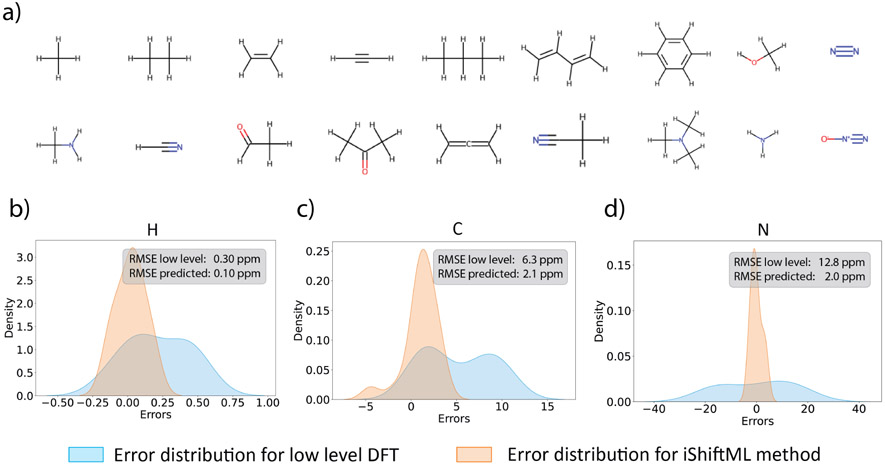
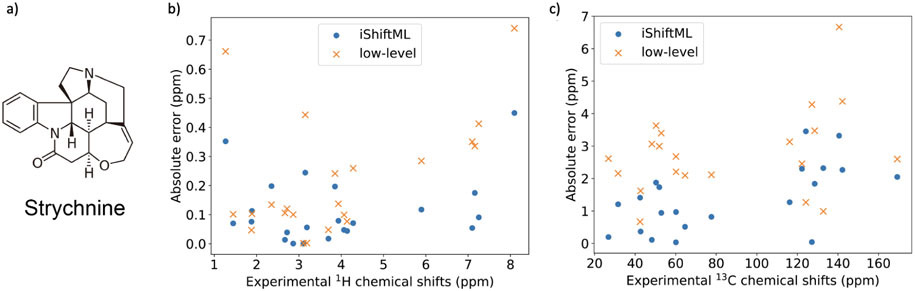
Theoretical predictions of NMR chemical shifts from first-principles can greatly facilitate experimental interpretation and structure identification of molecules in gas, solution, and solid-state phases. However, accurate prediction of chemical shifts using the gold-standard coupled cluster with singles, doubles, and perturbative triple excitations [CCSD(T)] method with a complete basis set (CBS) can be prohibitively expensive. By contrast, machine learning (ML) methods offer inexpensive alternatives for chemical shift predictions but are hampered by generalization to molecules outside the original training set. Here, we propose several new ideas in ML of the chemical shift prediction for H, C, N, and O that first introduce a novel feature representation, based on the atomic chemical shielding tensors within a molecular environment using an inexpensive quantum mechanics (QM) method, and train it to predict NMR chemical shieldings of a high-level composite theory that approaches the accuracy of CCSD(T)/CBS. In addition, we train the ML model through a new progressive active learning workflow that reduces the total number of expensive high-level composite calculations required while allowing the model to continuously improve on unseen data. Furthermore, the algorithm provides an error estimation, signaling potential unreliability in predictions if the error is large. Finally, we introduce a novel approach to keep the rotational invariance of the features using tensor environment vectors (TEVs) that yields a ML model with the highest accuracy compared to a similar model using data augmentation. We illustrate the predictive capacity of the resulting inexpensive shift machine learning (iShiftML) models across several benchmarks, including unseen molecules in the NS372 data set, gas-phase experimental chemical shifts for small organic molecules, and much larger and more complex natural products in which we can accurately differentiate between subtle diastereomers based on chemical shift assignments.
Conflict of interest statement
DECLARATION OF INTERESTS
M.H.G. is a part-owner of Q-Chem Inc, whose software was used for many of the calculations reported here.
Figures

Similar articles
-
Why benchmark-quality computations are needed to reproduce 1-adamantyl cation NMR chemical shifts accurately.J Phys Chem A. 2011 Mar 24;115(11):2340-4. doi: 10.1021/jp1103356. Epub 2011 Mar 1. J Phys Chem A. 2011. PMID: 21361308
-
Electron correlation and vibrational effects in predictions of paramagnetic NMR shifts.Phys Chem Chem Phys. 2022 Jun 29;24(25):15230-15244. doi: 10.1039/d2cp01206e. Phys Chem Chem Phys. 2022. PMID: 35703010
-
Predicting Density Functional Theory-Quality Nuclear Magnetic Resonance Chemical Shifts via Δ-Machine Learning.J Chem Theory Comput. 2021 Feb 9;17(2):826-840. doi: 10.1021/acs.jctc.0c00979. Epub 2021 Jan 11. J Chem Theory Comput. 2021. PMID: 33428408
-
Calculations on noncovalent interactions and databases of benchmark interaction energies.Acc Chem Res. 2012 Apr 17;45(4):663-72. doi: 10.1021/ar200255p. Epub 2012 Jan 6. Acc Chem Res. 2012. PMID: 22225511 Review.
-
Transferability Across Different Molecular Systems and Levels of Theory with the Data-Driven Coupled-Cluster Scheme.J Phys Chem A. 2025 Apr 3;129(13):2988-2997. doi: 10.1021/acs.jpca.4c05718. Epub 2025 Mar 25. J Phys Chem A. 2025. PMID: 40132101 Review.
Cited by
-
UCBShift 2.0: Bridging the Gap from Backbone to Side Chain Protein Chemical Shift Prediction for Protein Structures.J Am Chem Soc. 2024 Nov 20;146(46):31733-31745. doi: 10.1021/jacs.4c10474. Epub 2024 Nov 12. J Am Chem Soc. 2024. PMID: 39531038 Free PMC article.
-
Accurate Prediction of NMR Chemical Shifts: Integrating DFT Calculations with Three-Dimensional Graph Neural Networks.J Chem Theory Comput. 2024 Jun 25;20(12):5250-5258. doi: 10.1021/acs.jctc.4c00422. Epub 2024 Jun 6. J Chem Theory Comput. 2024. PMID: 38842505 Free PMC article.
-
Accurate and Efficient Structure Elucidation from Routine One-Dimensional NMR Spectra Using Multitask Machine Learning.ACS Cent Sci. 2024 Nov 13;10(11):2162-2170. doi: 10.1021/acscentsci.4c01132. eCollection 2024 Nov 27. ACS Cent Sci. 2024. PMID: 39634219 Free PMC article.
-
The interplay of density functional selection and crystal structure for accurate NMR chemical shift predictions.Faraday Discuss. 2025 Jan 8;255(0):119-142. doi: 10.1039/d4fd00072b. Faraday Discuss. 2025. PMID: 39258864
-
Bent naphthodithiophenes: synthesis and characterization of isomeric fluorophores.RSC Adv. 2024 Aug 12;14(35):25120-25129. doi: 10.1039/d4ra04850d. eCollection 2024 Aug 12. RSC Adv. 2024. PMID: 39139244 Free PMC article.
References
-
- Jacobsen NE NMR data interpretation explained: understanding 1D and 2D NMR spectra of organic compounds and natural products; John Wiley & Sons, 2016.
-
- Hore PJ Nuclear magnetic resonance; Oxford University Press, USA, 2015.
-
- Derome AE Modern NMR techniques for chemistry research; Elsevier, 2013.
-
- Wüthrich K. Protein structure determination in solution by NMR spectroscopy. J. Biol. Chem 1990, 265, 22059–22062. - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources