

APOE traffics to astrocyte lipid droplets and modulates triglyceride saturation and droplet size
- PMID: 38334983
- PMCID: PMC10857907
- DOI: 10.1083/jcb.202305003
APOE traffics to astrocyte lipid droplets and modulates triglyceride saturation and droplet size
Abstract
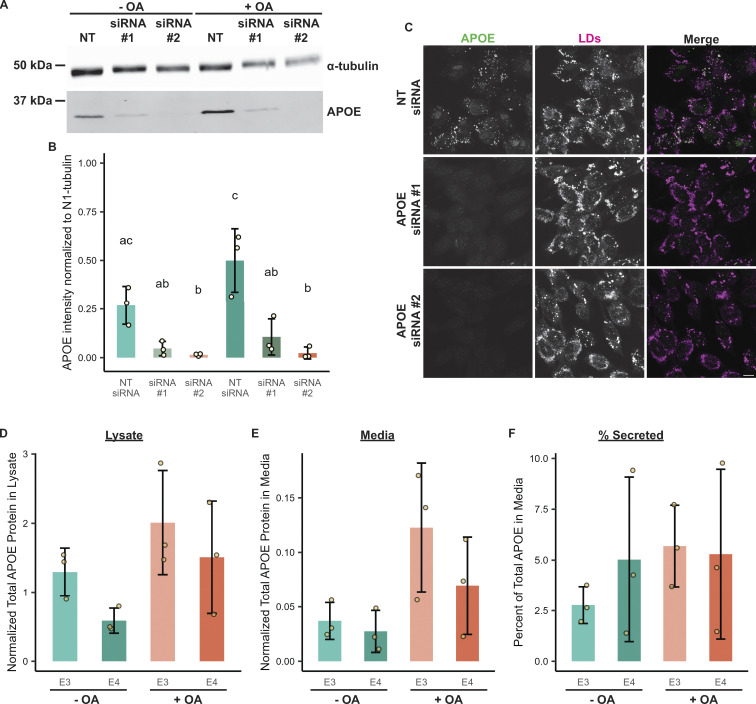
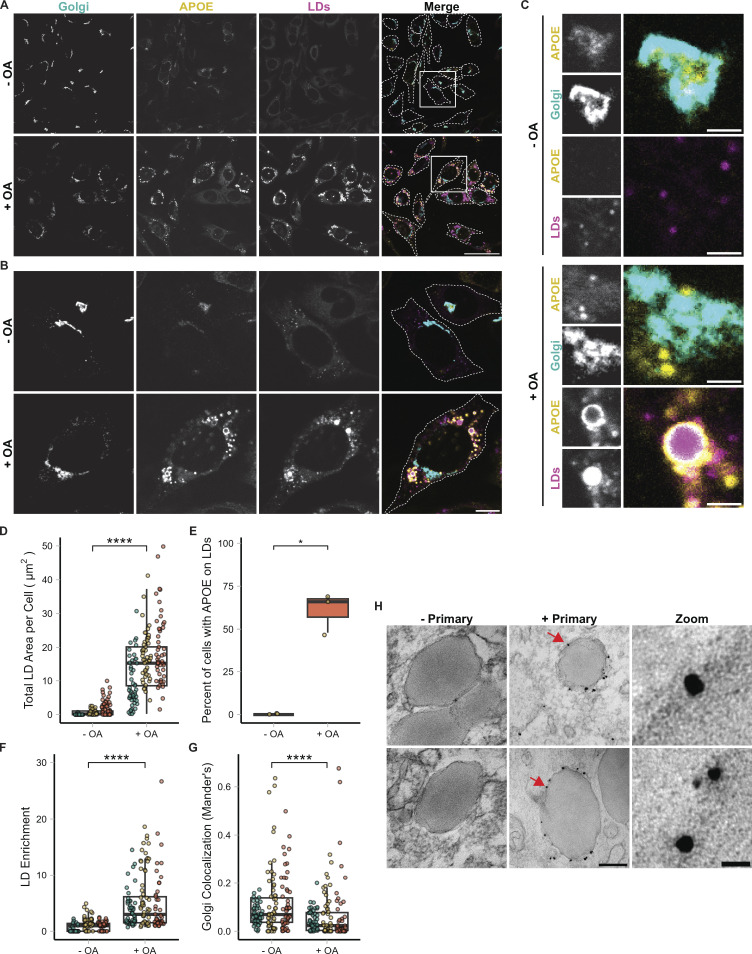
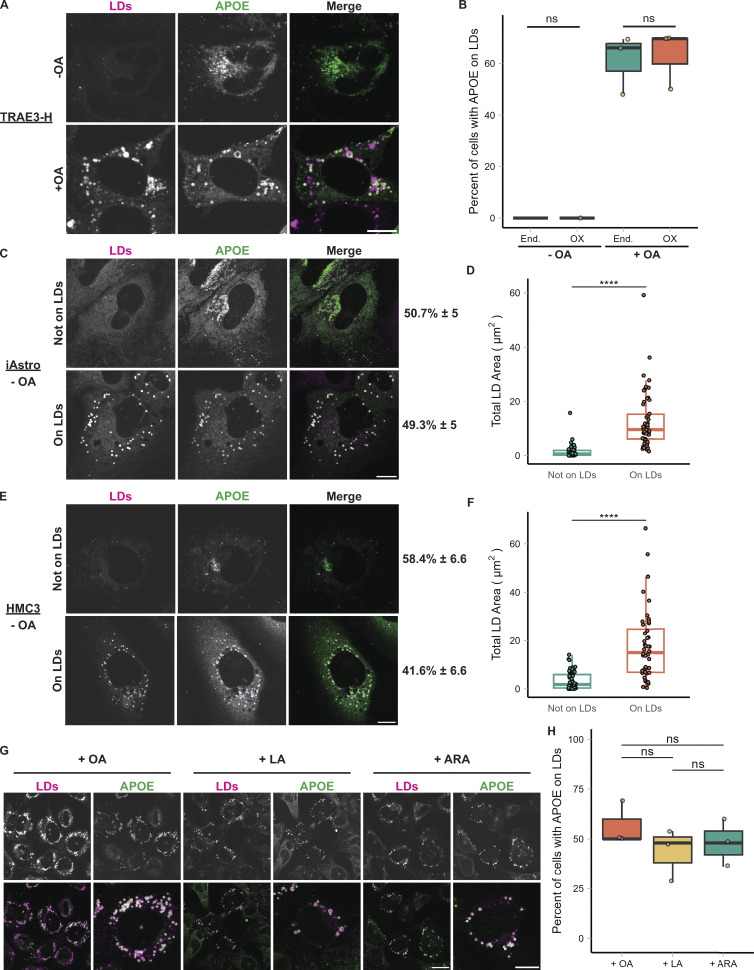
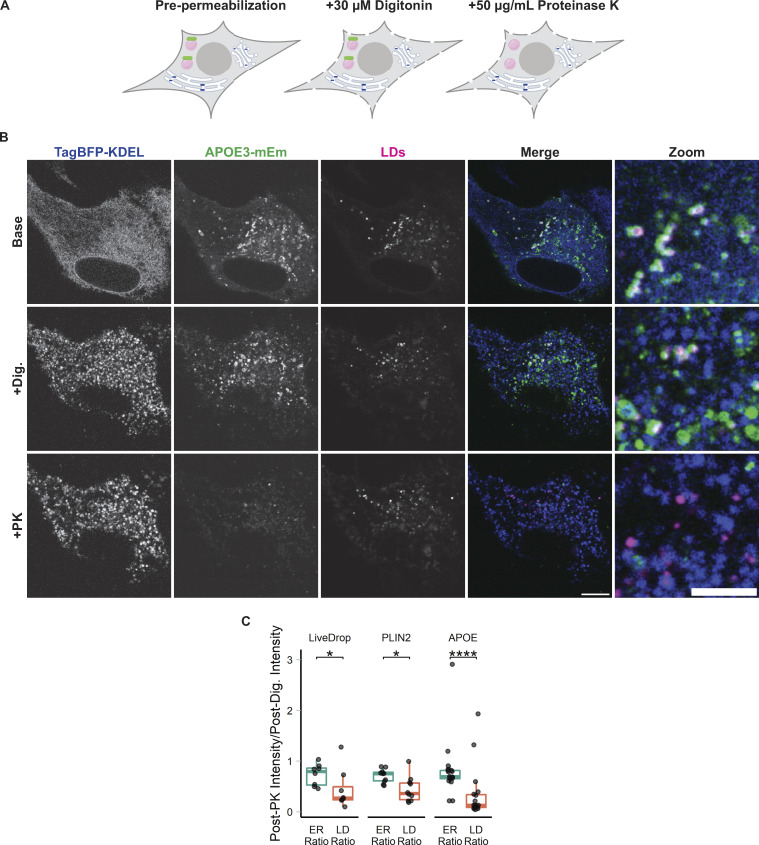
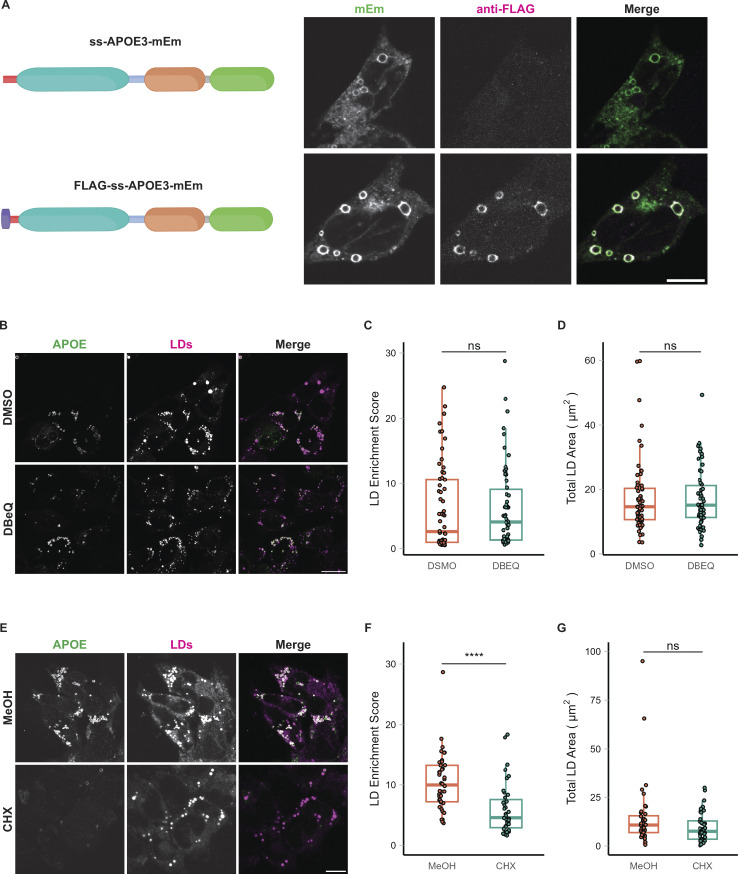
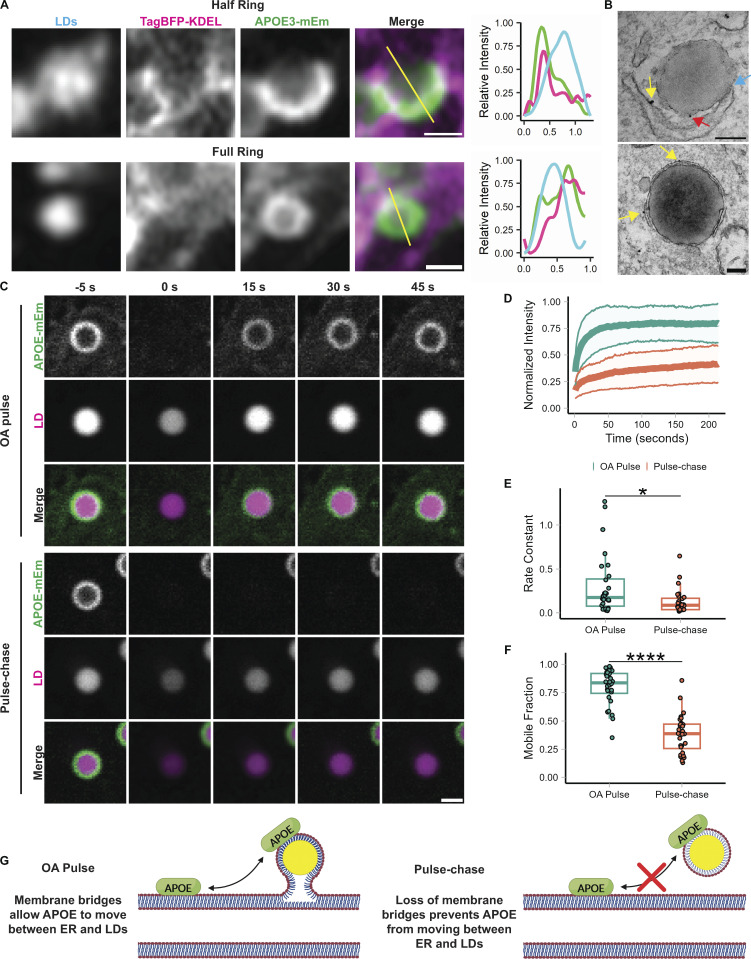
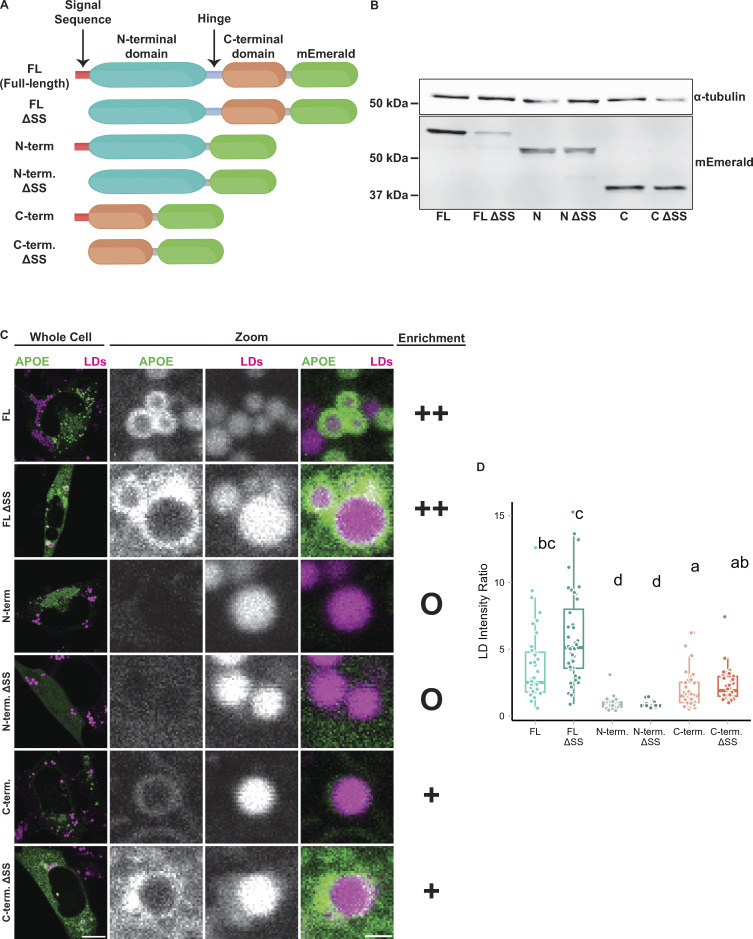
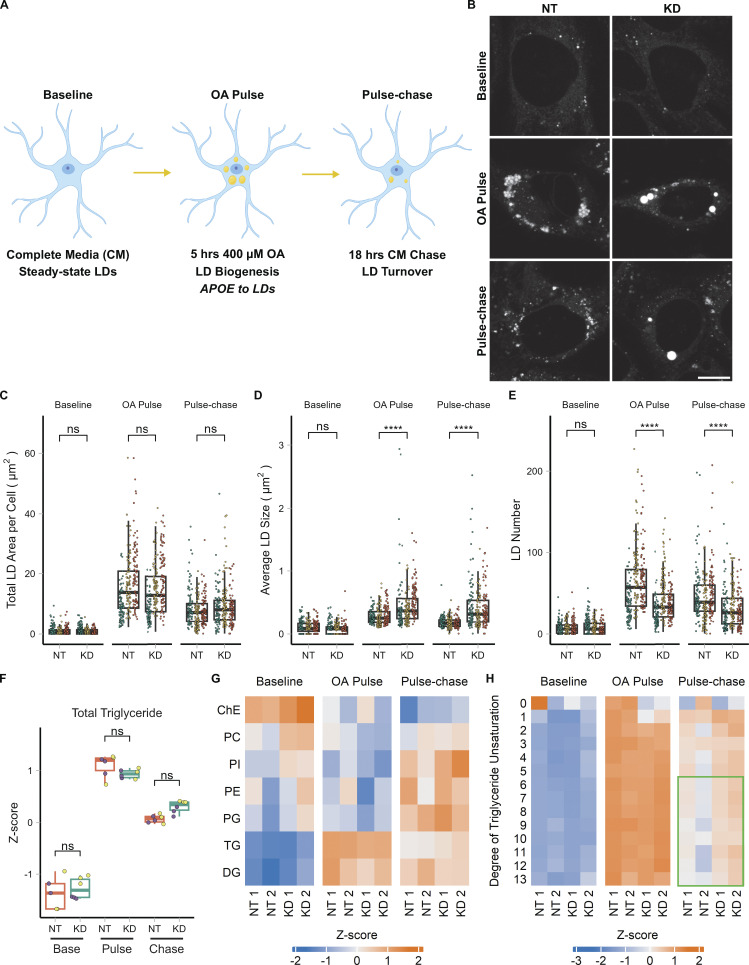
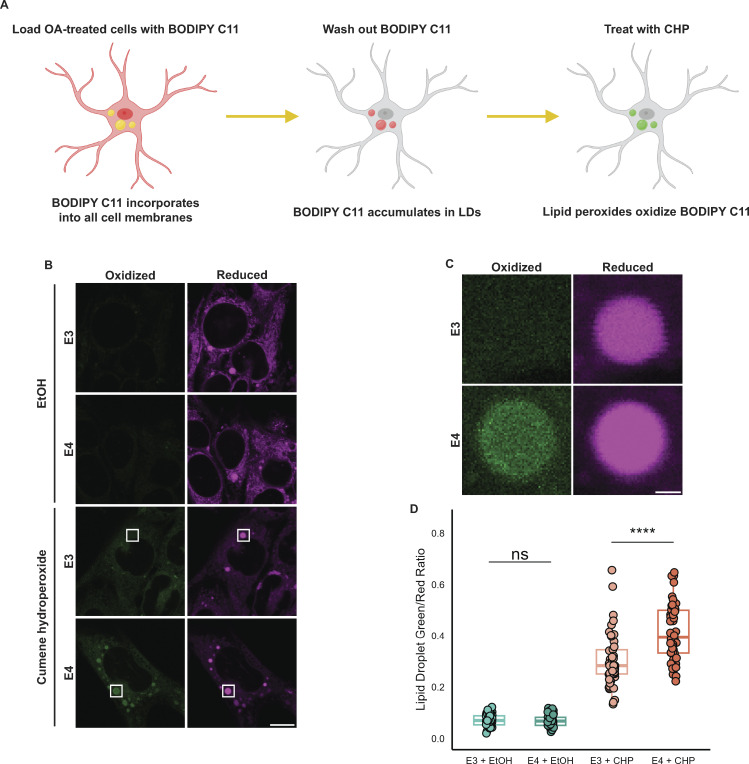
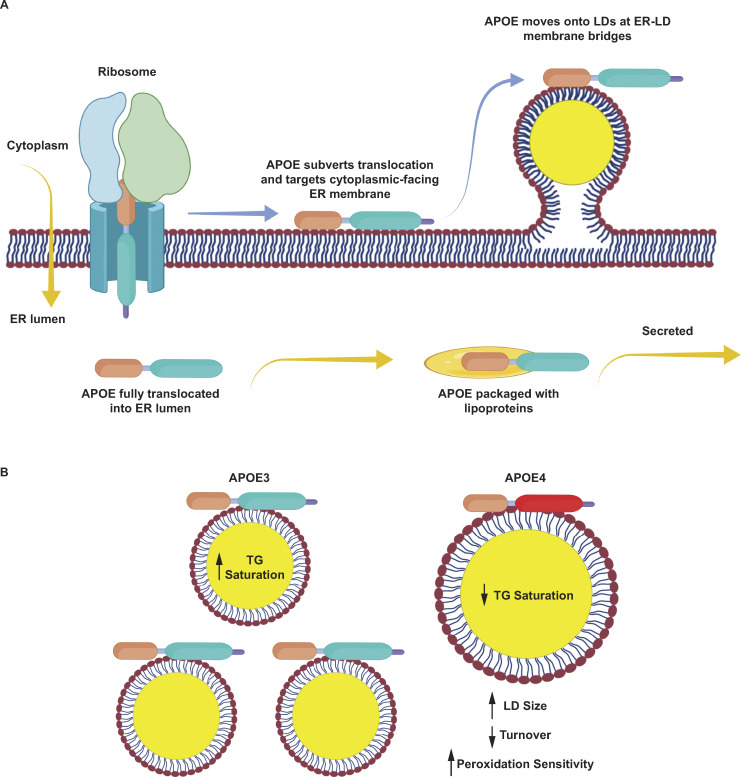
The E4 variant of APOE strongly predisposes individuals to late-onset Alzheimer's disease. We demonstrate that in response to lipogenesis, apolipoprotein E (APOE) in astrocytes can avoid translocation into the endoplasmic reticulum (ER) lumen and traffic to lipid droplets (LDs) via membrane bridges at ER-LD contacts. APOE knockdown promotes fewer, larger LDs after a fatty acid pulse, which contain more unsaturated triglyceride after fatty acid pulse-chase. This LD size phenotype was rescued by chimeric APOE that targets only LDs. Like APOE depletion, APOE4-expressing astrocytes form a small number of large LDs enriched in unsaturated triglyceride. Additionally, the LDs in APOE4 cells exhibit impaired turnover and increased sensitivity to lipid peroxidation. Our data indicate that APOE plays a previously unrecognized role as an LD surface protein that regulates LD size and composition. APOE4 causes aberrant LD composition and morphology. Our study contributes to accumulating evidence that APOE4 astrocytes with large, unsaturated LDs are sensitized to lipid peroxidation, which could contribute to Alzheimer's disease risk.
© 2024 Windham et al.
Conflict of interest statement
Disclosures: The authors declare no competing interests exist.
Figures
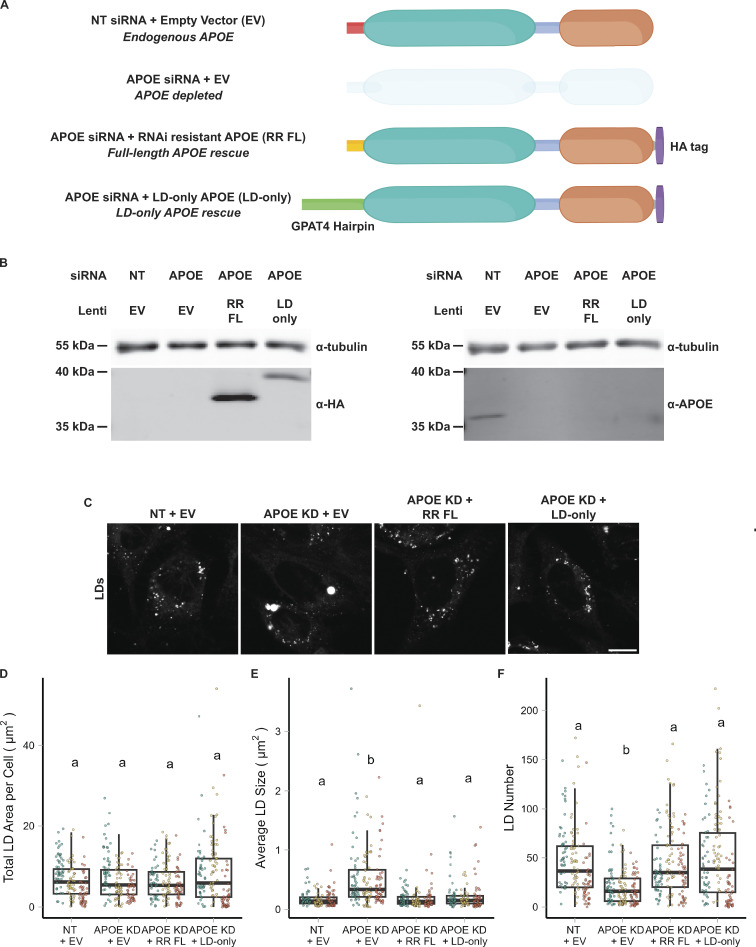
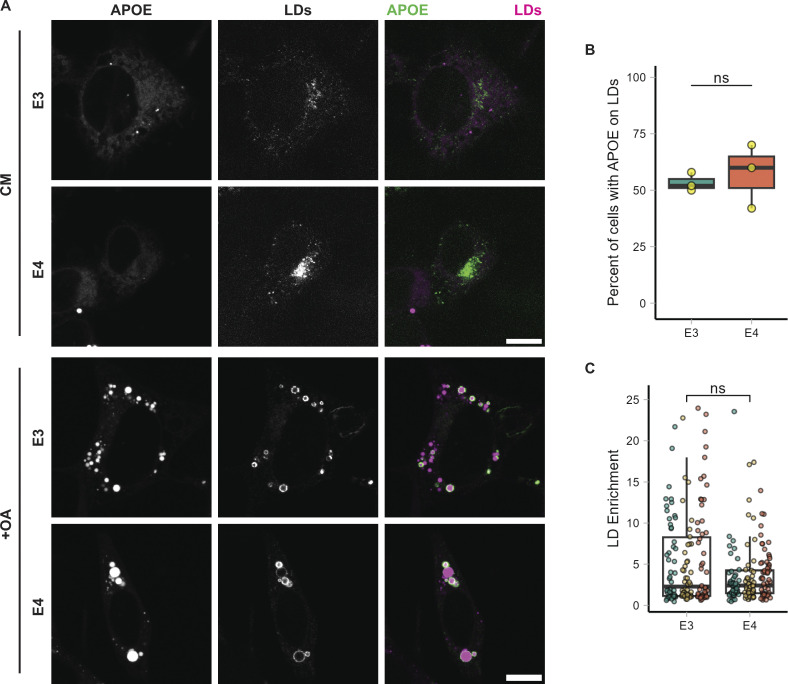
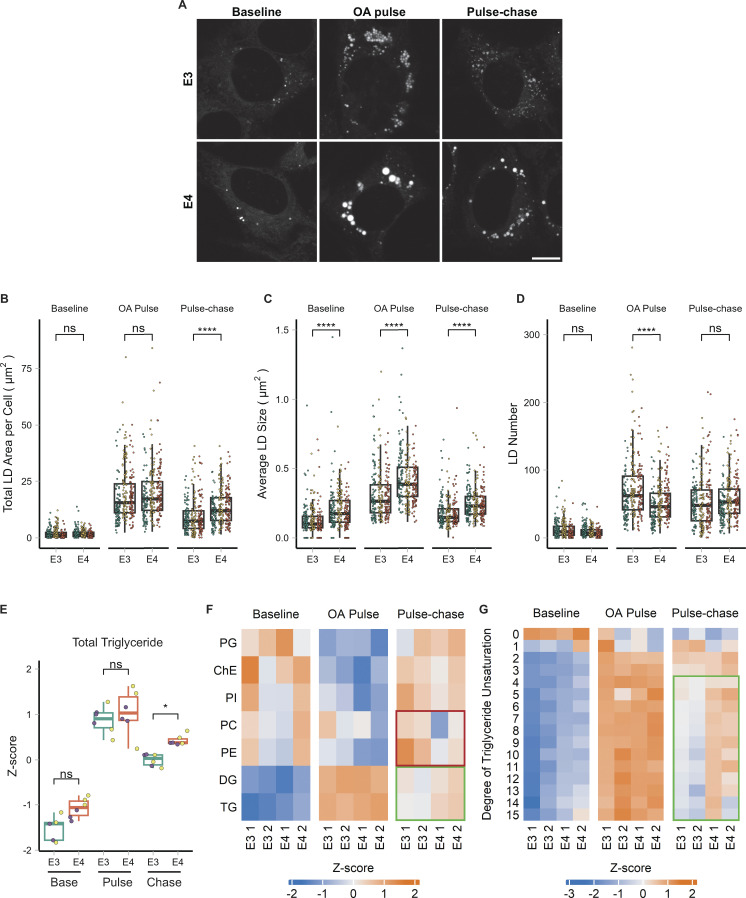
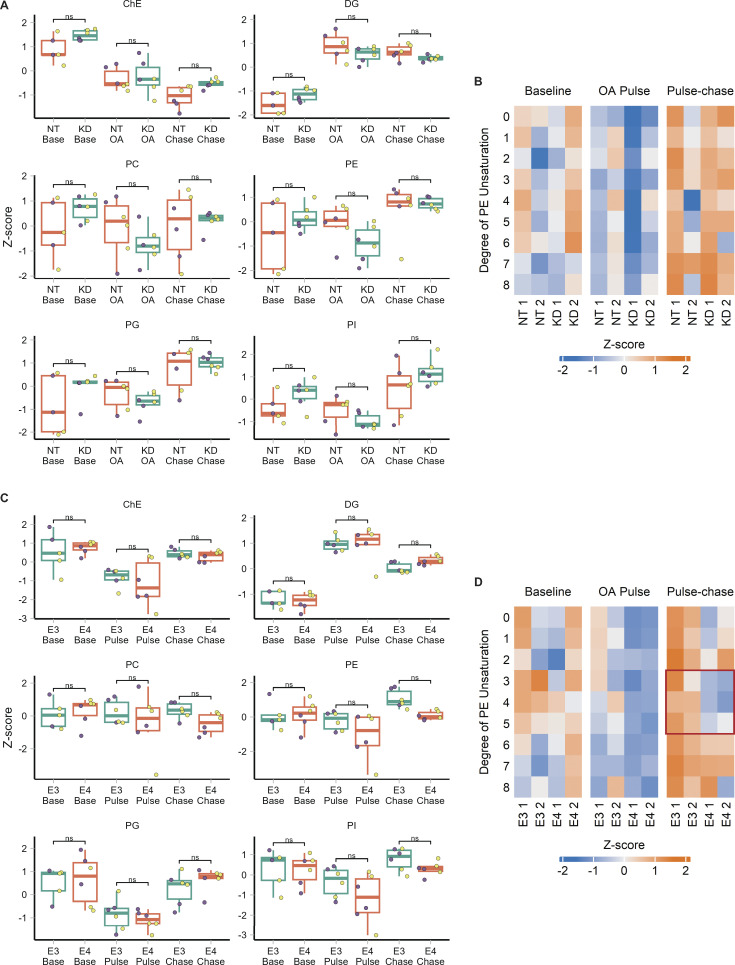
Update of
-
APOE traffics to astrocyte lipid droplets and modulates triglyceride saturation and droplet size.bioRxiv [Preprint]. 2023 Apr 29:2023.04.28.538740. doi: 10.1101/2023.04.28.538740. bioRxiv. 2023. Update in: J Cell Biol. 2024 Apr 1;223(4):e202305003. doi: 10.1083/jcb.202305003. PMID: 37162939 Free PMC article. Updated. Preprint.
References
-
- Alzheimer, A. 1907. Über Eine eigenartige Erkrankung der Hirnrinde. Allgemeine Zeitschrift für Psychiatrie und psychisch-Gerichtliche Medizin. 64:146–148.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
