

Identification of Hypoxia Prognostic Signature in Glioblastoma Multiforme Based on Bulk and Single-Cell RNA-Seq
- PMID: 38339384
- PMCID: PMC10854729
- DOI: 10.3390/cancers16030633
Identification of Hypoxia Prognostic Signature in Glioblastoma Multiforme Based on Bulk and Single-Cell RNA-Seq
Abstract
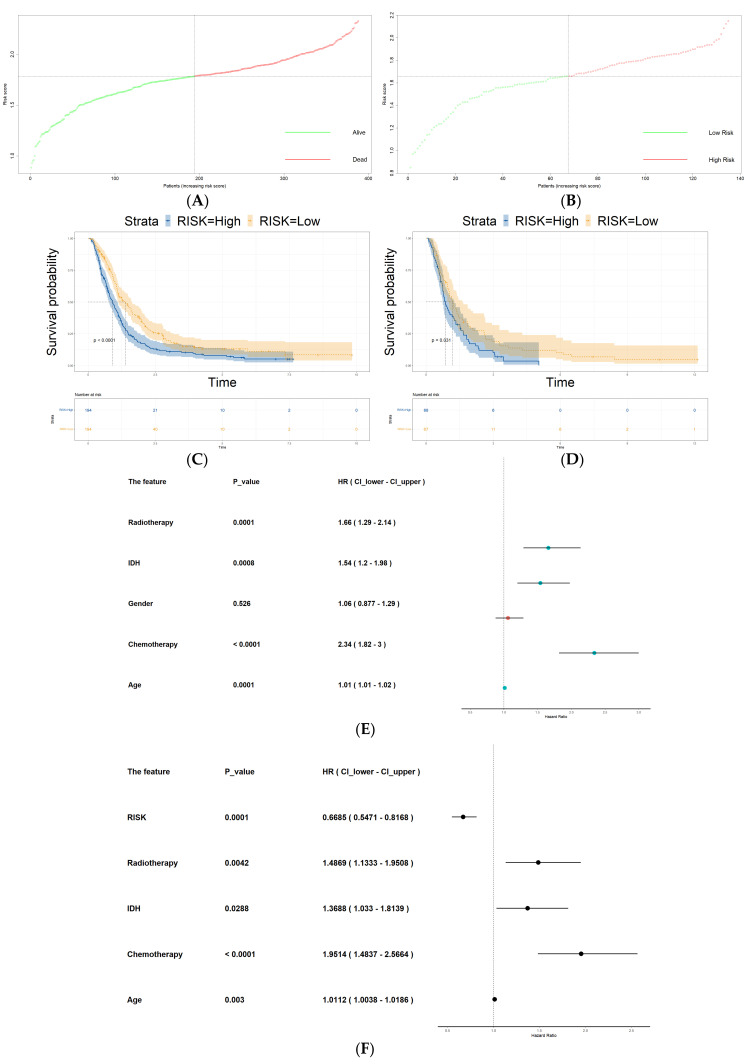
Glioblastoma (GBM) represents a profoundly aggressive and heterogeneous brain neoplasm linked to a bleak prognosis. Hypoxia, a common feature in GBM, has been linked to tumor progression and therapy resistance. In this study, we aimed to identify hypoxia-related differentially expressed genes (DEGs) and construct a prognostic signature for GBM patients using multi-omics analysis. Patient cohorts were collected from publicly available databases, including the Gene Expression Omnibus (GEO), the Chinese Glioma Genome Atlas (CGGA), and The Cancer Genome Atlas-Glioblastoma Multiforme (TCGA-GBM), to facilitate a comprehensive analysis. Hypoxia-related genes (HRGs) were obtained from the Molecular Signatures Database (MSigDB). Differential expression analysis revealed 41 hypoxia-related DEGs in GBM patients. A consensus clustering approach, utilizing these DEGs' expression patterns, identified four distinct clusters, with cluster 1 showing significantly better overall survival. Machine learning techniques, including univariate Cox regression and LASSO regression, delineated a prognostic signature comprising six genes (ANXA1, CALD1, CP, IGFBP2, IGFBP5, and LOX). Multivariate Cox regression analysis substantiated the prognostic significance of a set of three optimal signature genes (CP, IGFBP2, and LOX). Using the hypoxia-related prognostic signature, patients were classified into high- and low-risk categories. Survival analysis demonstrated that the high-risk group exhibited inferior overall survival rates in comparison to the low-risk group. The prognostic signature showed good predictive performance, as indicated by the area under the curve (AUC) values for one-, three-, and five-year overall survival. Furthermore, functional enrichment analysis of the DEGs identified biological processes and pathways associated with hypoxia, providing insights into the underlying mechanisms of GBM. Delving into the tumor immune microenvironment, our analysis revealed correlations relating the hypoxia-related prognostic signature to the infiltration of immune cells in GBM. Overall, our study highlights the potential of a hypoxia-related prognostic signature as a valuable resource for forecasting the survival outcome of GBM patients. The multi-omics approach integrating bulk sequencing, single-cell analysis, and immune microenvironment assessment enhances our understanding of the intricate biology characterizing GBM, thereby potentially informing the tailored design of therapeutic interventions.
Keywords: CP; IGFBP2; LOX; bioinformatics; glioblastoma multiforme; hypoxia.
Conflict of interest statement
The authors declare no conflicts of interest.
Figures









References
-
- Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S., et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. - DOI - PMC - PubMed
-
- Winter S.C., Buffa F.M., Silva P., Miller C., Valentine H.R., Turley H., Shah K.A., Cox G.J., Corbridge R.J., Homer J.J., et al. Relation of a Hypoxia Metagene Derived from Head and Neck Cancer to Prognosis of Multiple Cancers. Cancer Res. 2007;67:3441–3449. doi: 10.1158/0008-5472.CAN-06-3322. - DOI - PubMed
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
