

Radiotherapy-Induced Astrocyte Senescence Promotes an Immunosuppressive Microenvironment in Glioblastoma to Facilitate Tumor Regrowth
- PMID: 38342629
- PMCID: PMC11022718
- DOI: 10.1002/advs.202304609
Radiotherapy-Induced Astrocyte Senescence Promotes an Immunosuppressive Microenvironment in Glioblastoma to Facilitate Tumor Regrowth
Abstract
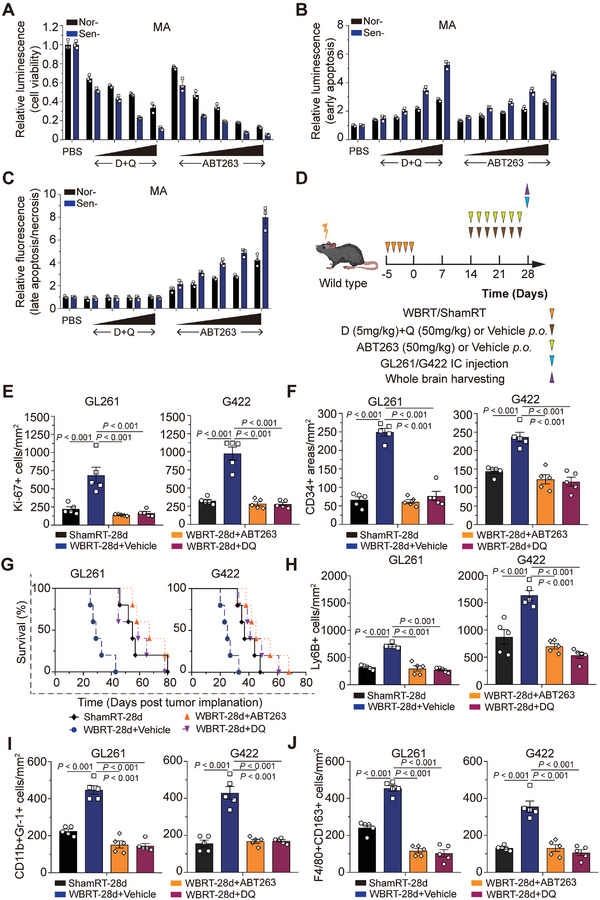
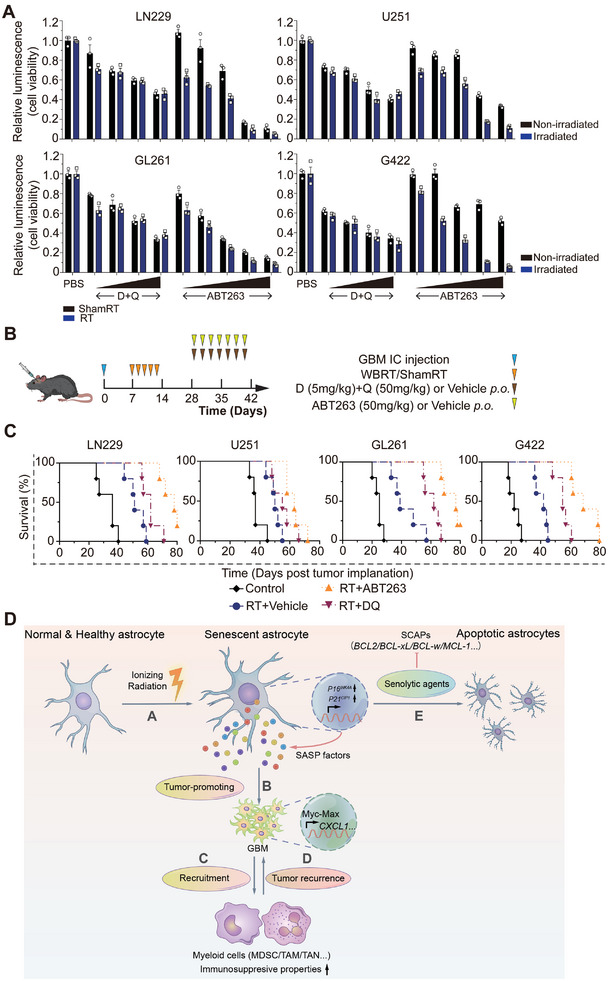
Accumulating evidence suggests that changes in the tumor microenvironment caused by radiotherapy are closely related to the recurrence of glioma. However, the mechanisms by which such radiation-induced changes are involved in tumor regrowth have not yet been fully investigated. In the present study, how cranial irradiation-induced senescence in non-neoplastic brain cells contributes to glioma progression is explored. It is observed that senescent brain cells facilitated tumor regrowth by enhancing the peripheral recruitment of myeloid inflammatory cells in glioblastoma. Further, it is identified that astrocytes are one of the most susceptible senescent populations and that they promoted chemokine secretion in glioma cells via the senescence-associated secretory phenotype. By using senolytic agents after radiotherapy to eliminate these senescent cells substantially prolonged survival time in preclinical models. The findings suggest the tumor-promoting role of senescent astrocytes in the irradiated glioma microenvironment and emphasize the translational relevance of senolytic agents for enhancing the efficacy of radiotherapy in gliomas.
Keywords: Glioma; astrocytes; myeloid inflammatory cells; radiation‐induced senescence; senolytic agents.
© 2024 The Authors. Advanced Science published by Wiley‐VCH GmbH.
Conflict of interest statement
The authors declare no conflict of interest.
Figures






References
-
- a) Stupp R., Hegi M. E., Mason W. P., van den M. J., Bent M. J., Taphoorn R. C., Janzer S. K., Ludwin A., Allgeier B., Fisher K., Belanger P., Hau A. A., Brandes J., Gijtenbeek C., Marosi C. J., Vecht K., Mokhtari P., Wesseling S., Villa E., Eisenhauer T., Gorlia M., Weller D., Lacombe J. G., Cairncross R. O., Mirimanoff R., Lancet Oncol. 2009, 10, 459,; - PubMed
- b) Sulman E. P., Ismaila N., Armstrong T. S., Tsien C., Batchelor T. T., Cloughesy T., Galanis E., Gilbert M., Gondi V., Lovely M., Mehta M., Mumber M. P., Sloan A., Chang S. M., J. Clin. Oncol. 2017, 35, 361. - PubMed
-
- Wernicke A. G., Smith A. W., Taube S., Mehta M. P., Pract Radiat Oncol 2016, 6, 298. - PubMed
-
- Jiang W., Chan C. K., Weissman I. L., Kim B. Y. S., Hahn S. M., Trends Cancer 2016, 2, 638. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases