

DNA methylation and transcriptome analysis reveal epigenomic differences among three macaque species
- PMID: 38343783
- PMCID: PMC10853583
- DOI: 10.1111/eva.13604
DNA methylation and transcriptome analysis reveal epigenomic differences among three macaque species
Abstract
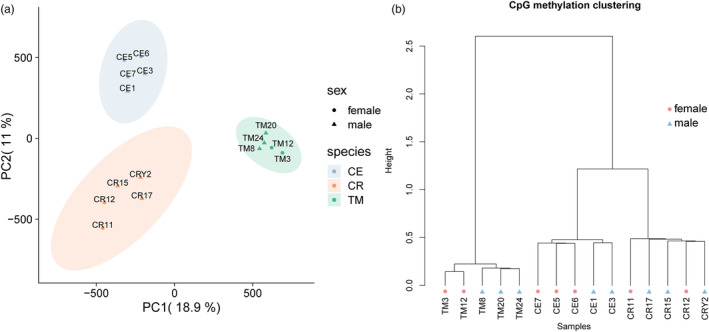
Macaques (genus Macaca) are the most widely distributed non-human primates, and their evolutionary history, gene expression profiles, and genetic differences have been extensively studied. However, the DNA methylomes of macaque species are not available in public databases, which hampers understanding of epigenetic differences among macaque species. Epigenetic modifications can potentially affect development, physiology, behavior, and evolution. Here, we investigated the methylation patterns of the Tibetan macaque (M. thibetana; TM), Chinese rhesus macaque (M. mulatta lasiota; CR), and crab-eating macaque (M. fascicularis; CE) through whole-genome bisulfite sequencing from peripheral blood. We compared genome-wide methylation site information for the three species. We identified 12,128 (CR vs. CE), 59,165 (CR vs. TM), and 39,751 (CE vs. TM) differentially methylated regions (DMRs) in the three macaques. Furthermore, we obtained the differentially expressed genes (DEGs) among the three macaque species. The differences between CR and CE were smaller at both the methylome and transcriptome levels than compared with TM (CR vs. TM and CE vs. TM). We also found a change in the density of single nucleotide mutations in DMRs relative to their flanking regions, indicating a potential mechanism through which genomic alterations may modulate methylation landscapes, thereby influencing the transcriptome. Functional enrichment analyses showed the DMR-related genes were enriched in developmental processes and neurological functions, such as the growth hormone-related pathway, insulin secretion pathway, thyroid hormone synthesis pathway, morphine addiction, and GABAergic synapses. These differences may be associated with variations in physiology and habitat among the macaques. Our study provides one of the first genome-wide comparisons of genetic, gene expression, and epigenetic variations across different macaques. Our results should facilitate further research on comparative genomic and genetic differences in macaque species.
Keywords: DNA methylation; Macaca; epigenetics; gene expression; neuro development.
© 2023 The Authors. Evolutionary Applications published by John Wiley & Sons Ltd.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures




References
-
- Angers, B. , Castonguay, E. , & Massicotte, R. (2010). Environmentally induced phenotypes and DNA methylation: How to deal with unpredictable conditions until the next generation and after. Molecular Ecology, 19(7), 1283–1295. - PubMed
-
- Bonkovsky, H. L. , Guo, J. T. , Hou, W. , Li, T. , Narang, T. , & Thapar, M. (2013). Porphyrin and heme metabolism and the porphyrias. Comprehensive Physiology, 3(1), 365–401. - PubMed
LinkOut - more resources
Full Text Sources
