

This is a preprint.
Host DNA depletion on frozen human respiratory samples enables successful metagenomic sequencing for microbiome studies
- PMID: 38343829
- PMCID: PMC10854296
- DOI: 10.21203/rs.3.rs-3638876/v1
Host DNA depletion on frozen human respiratory samples enables successful metagenomic sequencing for microbiome studies
Update in
-
Host DNA depletion on frozen human respiratory samples enables successful metagenomic sequencing for microbiome studies.Commun Biol. 2024 Nov 28;7(1):1590. doi: 10.1038/s42003-024-07290-3. Commun Biol. 2024. PMID: 39609616 Free PMC article.
Abstract
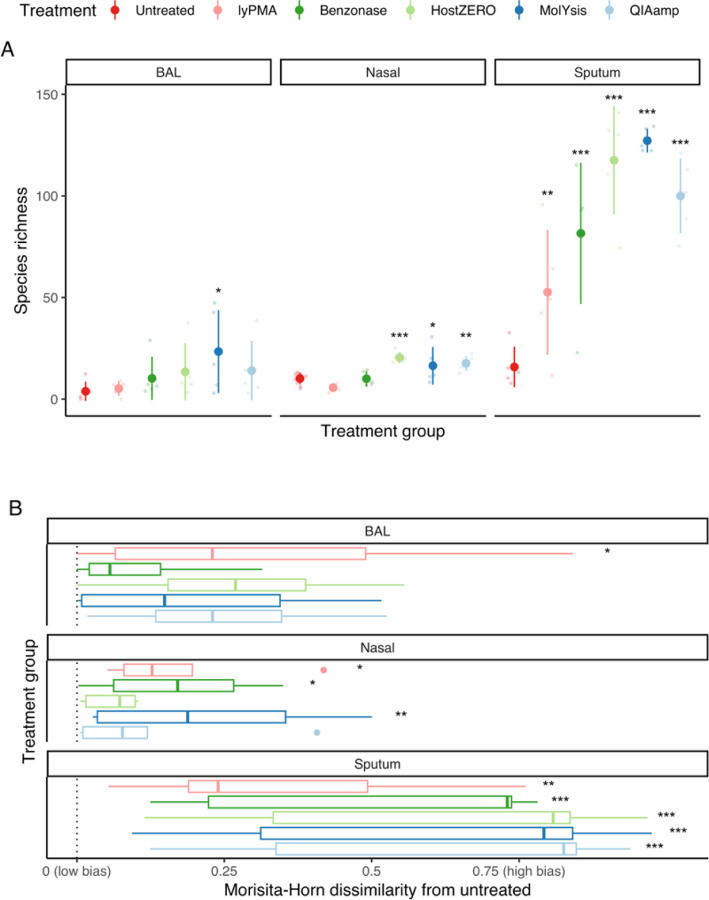
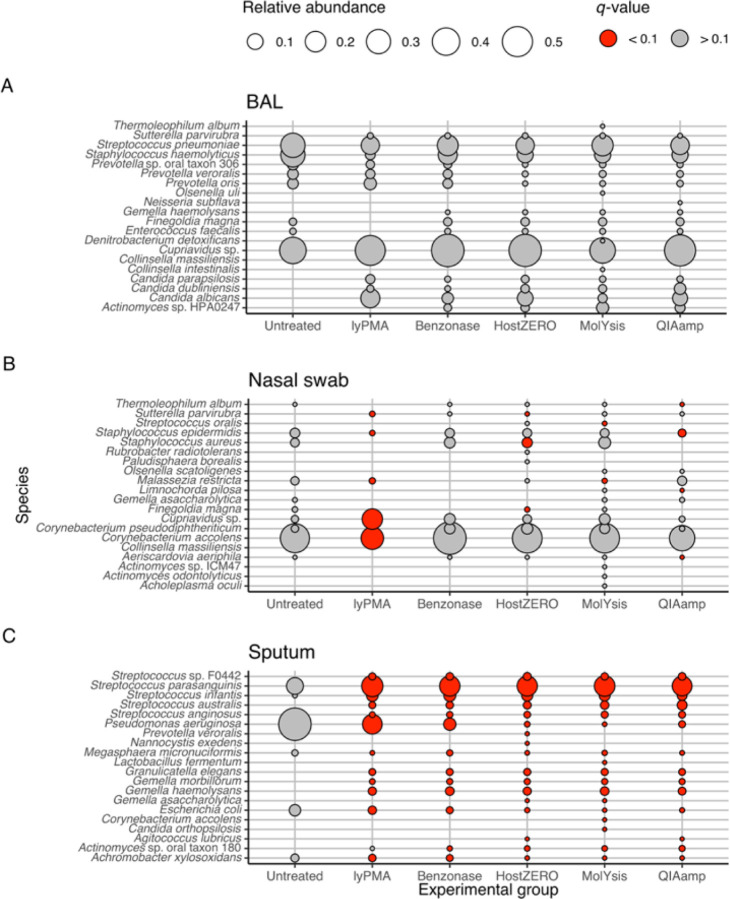
Background: Most respiratory microbiome studies have focused on amplicon rather than metagenomics sequencing due to high host DNA content. We evaluated efficacy of five host DNA depletion methods on previously frozen human bronchoalveolar lavage (BAL), nasal swabs, and sputum prior to metagenomic sequencing.
Results: Median sequencing depth was 76.4 million reads per sample. Untreated nasal, sputum and BAL samples had 94.1%, 99.2%, and 99.7% host-reads. The effect of host depletion differed by sample type. Most treatment methods increased microbial reads, species richness and predicted functional richness; the increase in species and predicted functional richness was mediated by higher effective sequencing depth. For BAL and nasal samples, most methods did not change Morisita-Horn dissimilarity suggesting limited bias introduced by host depletion.
Conclusions: Metagenomics sequencing without host depletion will underestimate microbial diversity of most respiratory samples due to shallow effective sequencing depth and is not recommended. Optimal host depletion methods vary by sample type.
Keywords: Microbiome; Respiratory; Shotgun metagenomics sequencing; host DNA depletion; low biomass.
Conflict of interest statement
Competing interests We declare the authors have no competing interests.
Figures


References
-
- Di Simone S. K., Rudloff I., Nold-Petry C. A., Forster S. C. & Nold M. F. Understanding respiratory microbiome-immune system interactions in health and disease. Sci. Transl. Med. 15, eabq5126 (2023). - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
