

Assessment of Two Restraint Potentials for Coarse-Grained Chemical-Cross-Link-Assisted Modeling of Protein Structures
- PMID: 38345917
- PMCID: PMC10900291
- DOI: 10.1021/acs.jcim.3c01890
Assessment of Two Restraint Potentials for Coarse-Grained Chemical-Cross-Link-Assisted Modeling of Protein Structures
Abstract
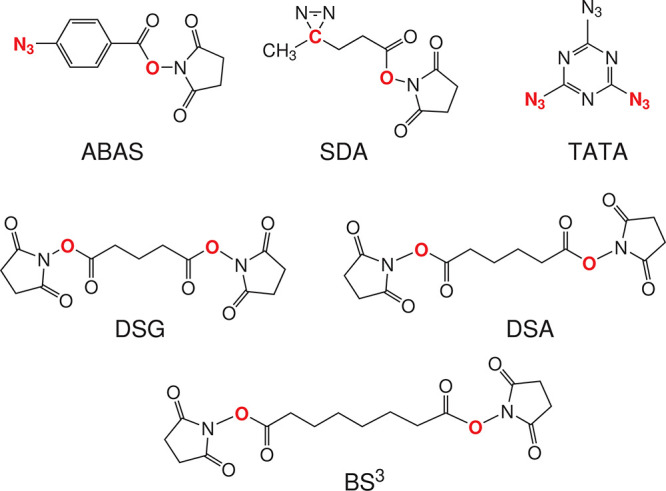
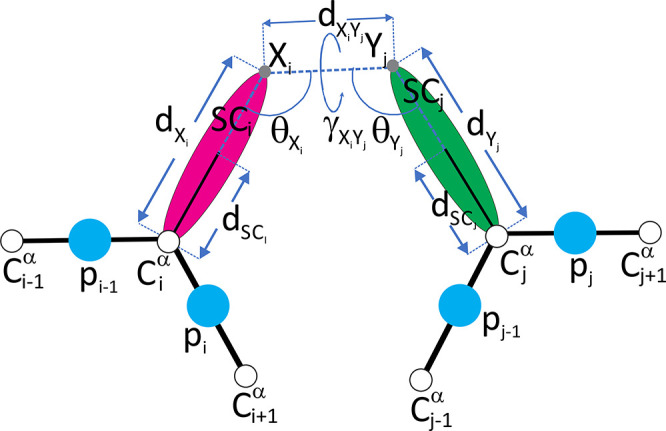
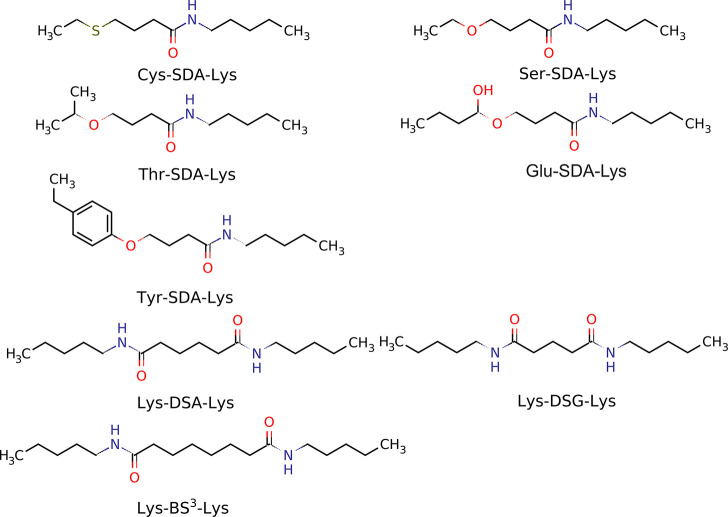
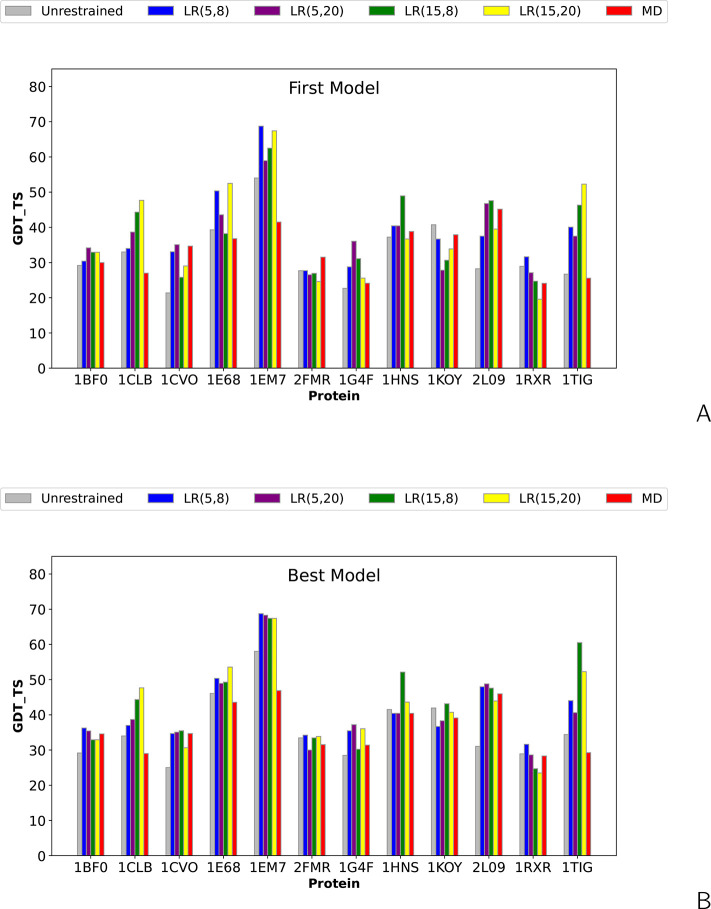
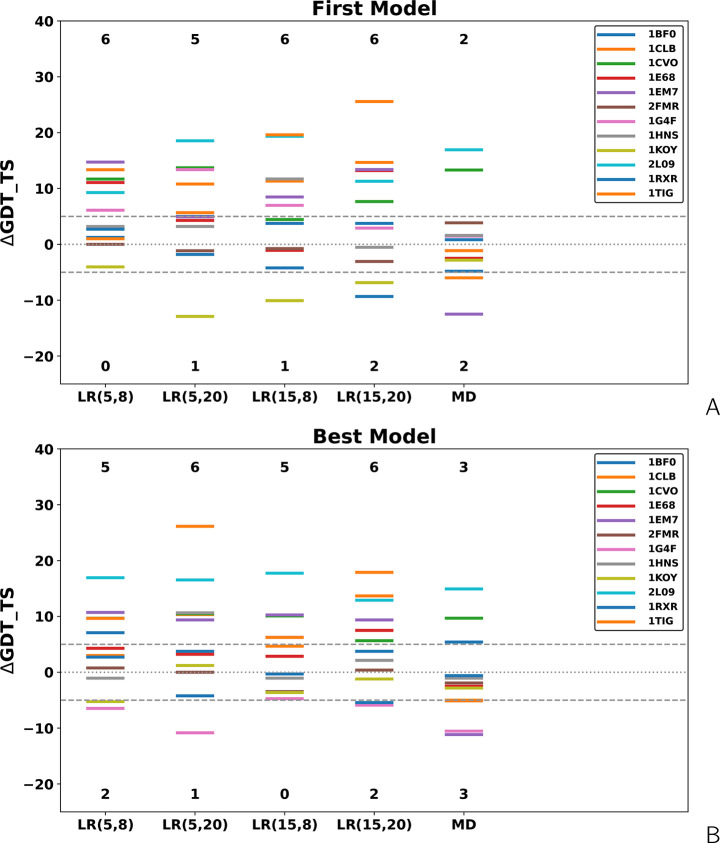
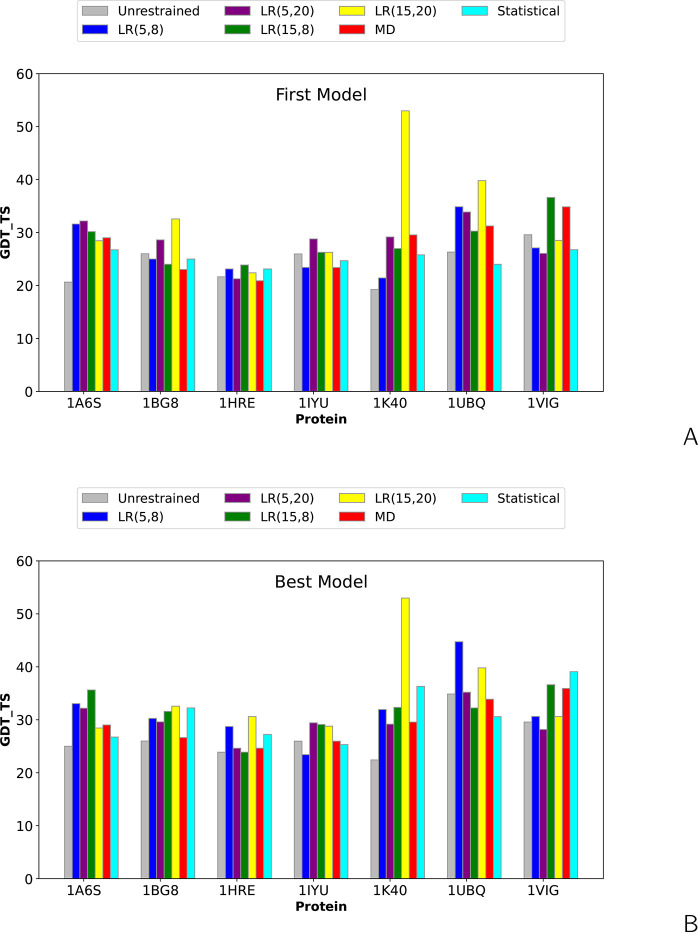
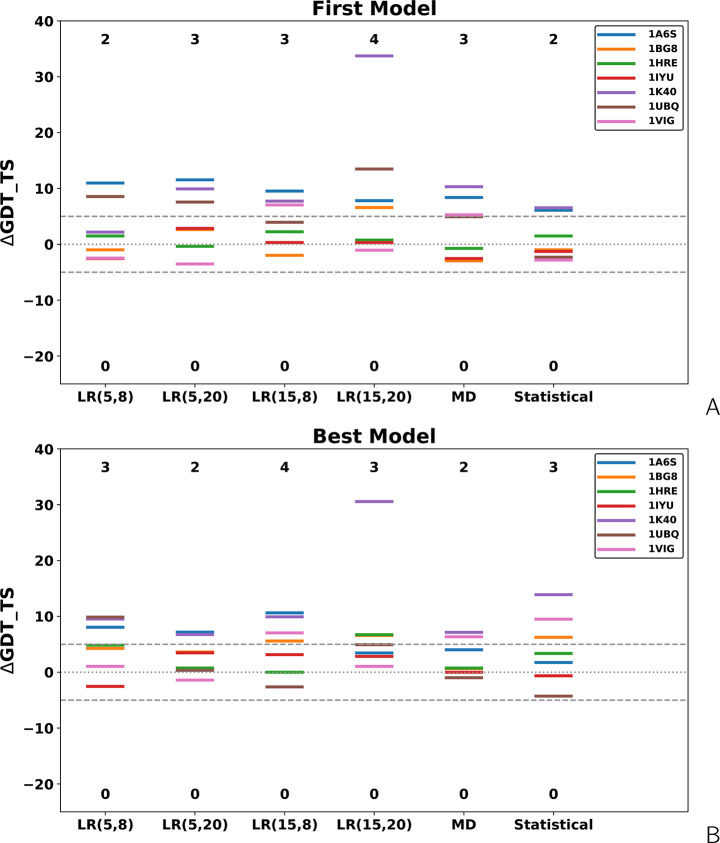
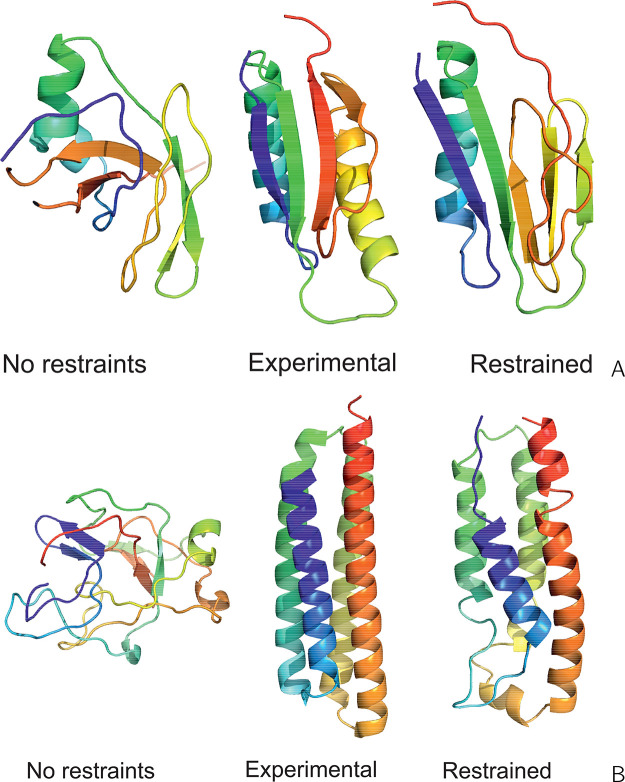
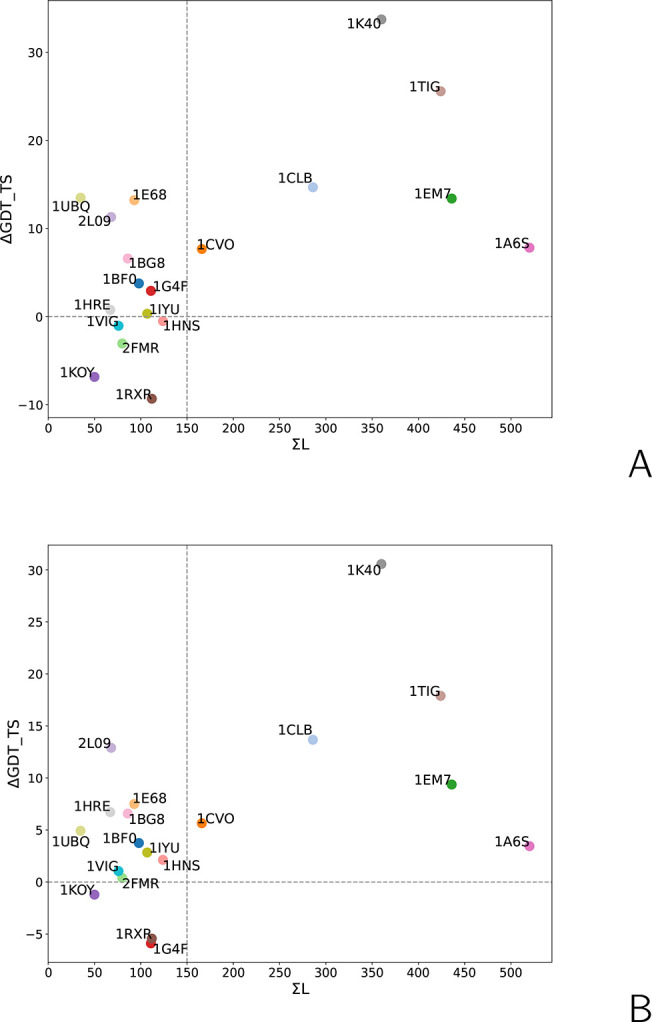
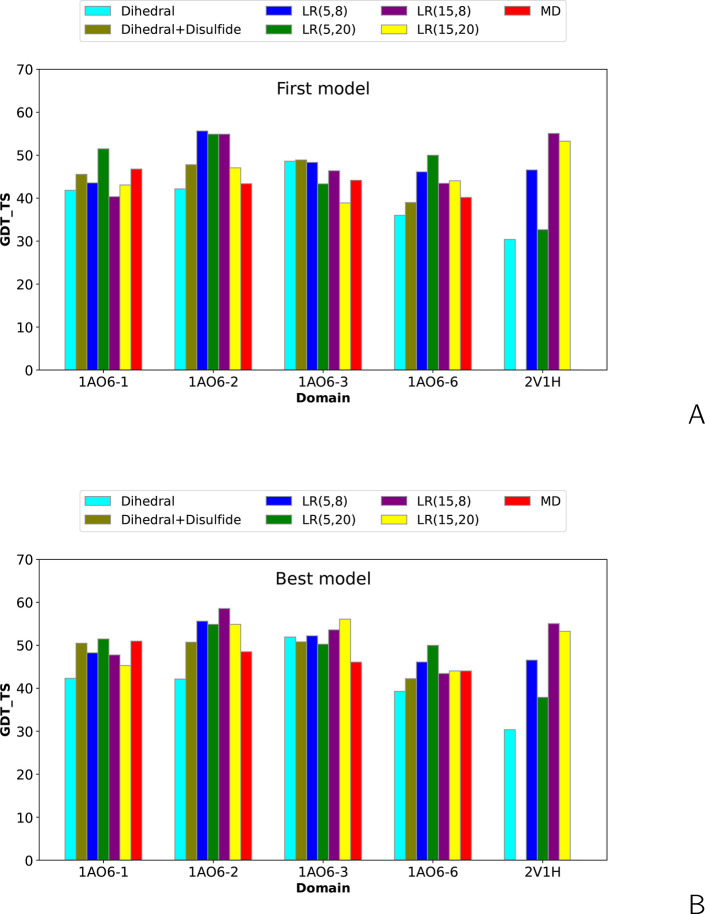
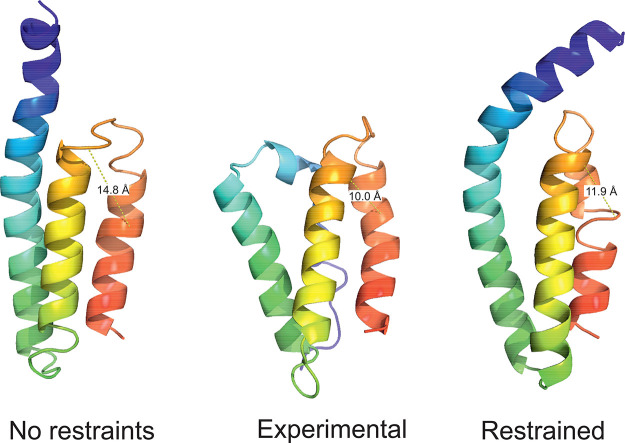
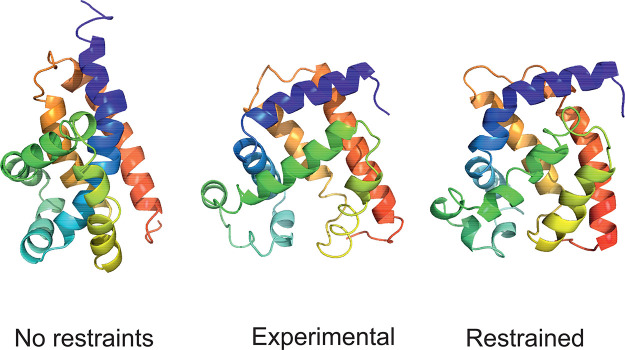
The influence of distance restraints from chemical cross-link mass spectroscopy (XL-MS) on the quality of protein structures modeled with the coarse-grained UNRES force field was assessed by using a protocol based on multiplexed replica exchange molecular dynamics, in which both simulated and experimental cross-link restraints were employed, for 23 small proteins. Six cross-links with upper distance boundaries from 4 Å to 12 Å (azido benzoic acid succinimide (ABAS), triazidotriazine (TATA), succinimidyldiazirine (SDA), disuccinimidyl adipate (DSA), disuccinimidyl glutarate (DSG), and disuccinimidyl suberate (BS3)) and two types of restraining potentials ((i) simple flat-bottom Lorentz-like potentials dependent on side chain distance (all cross-links) and (ii) distance- and orientation-dependent potentials determined based on molecular dynamics simulations of model systems (DSA, DSG, BS3, and SDA)) were considered. The Lorentz-like potentials with properly set parameters were found to produce a greater number of higher-quality models compared to unrestrained simulations than the MD-based potentials, because the latter can force too long distances between side chains. Therefore, the flat-bottom Lorentz-like potentials are recommended to represent cross-link restraints. It was also found that significant improvement of model quality upon the introduction of cross-link restraints is obtained when the sum of differences of indices of cross-linked residues exceeds 150.
Conflict of interest statement
The authors declare no competing financial interest.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
