

Predicting proximal tubule failed repair drivers through regularized regression analysis of single cell multiomic sequencing
- PMID: 38347009
- PMCID: PMC10861555
- DOI: 10.1038/s41467-024-45706-0
Predicting proximal tubule failed repair drivers through regularized regression analysis of single cell multiomic sequencing
Abstract
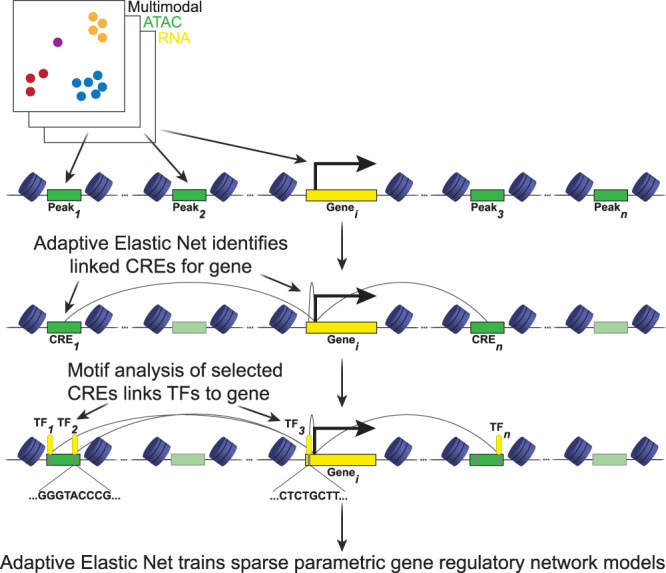
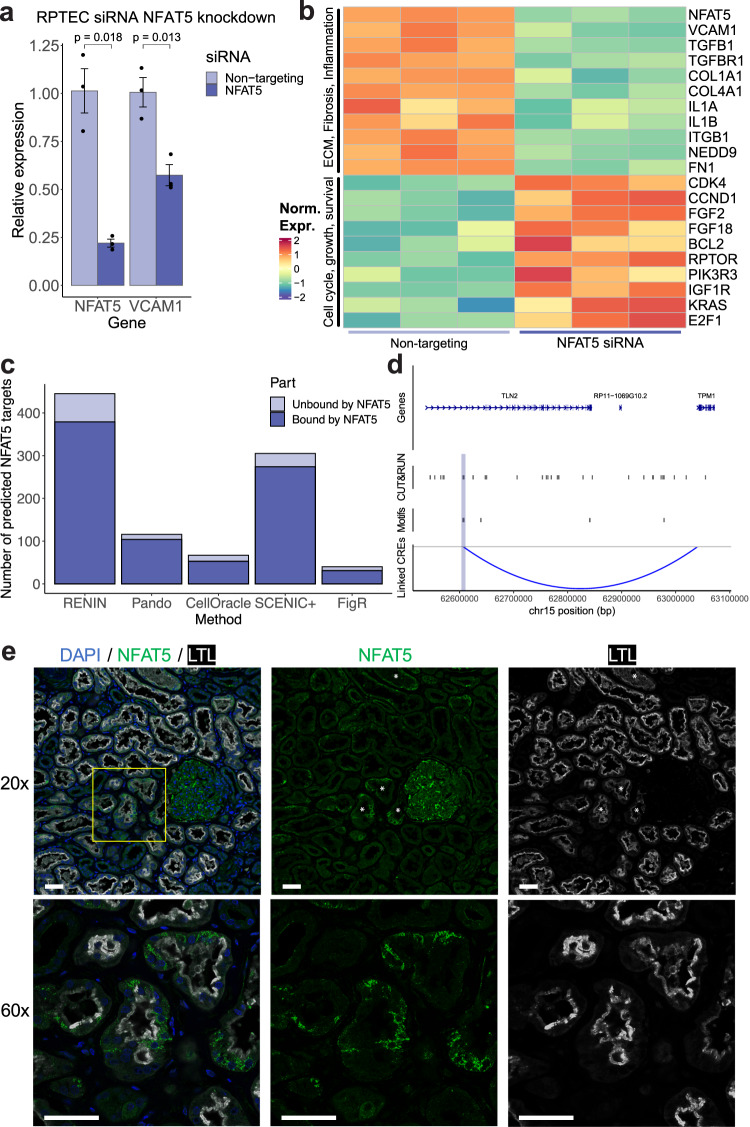
Renal proximal tubule epithelial cells have considerable intrinsic repair capacity following injury. However, a fraction of injured proximal tubule cells fails to undergo normal repair and assumes a proinflammatory and profibrotic phenotype that may promote fibrosis and chronic kidney disease. The healthy to failed repair change is marked by cell state-specific transcriptomic and epigenomic changes. Single nucleus joint RNA- and ATAC-seq sequencing offers an opportunity to study the gene regulatory networks underpinning these changes in order to identify key regulatory drivers. We develop a regularized regression approach to construct genome-wide parametric gene regulatory networks using multiomic datasets. We generate a single nucleus multiomic dataset from seven adult human kidney samples and apply our method to study drivers of a failed injury response associated with kidney disease. We demonstrate that our approach is a highly effective tool for predicting key cis- and trans-regulatory elements underpinning the healthy to failed repair transition and use it to identify NFAT5 as a driver of the maladaptive proximal tubule state.
© 2024. The Author(s).
Conflict of interest statement
B.D.H. is a consultant for Janssen Research & Development, LLC, Pfizer and Chinook Therapeutics, holds equity in Chinook Therapeutics and grant funding from Janssen Research & Development, LLC and Pfizer; all interests are unrelated to the current work.
Figures



References
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
