

Bioinformatic analysis of defective viral genomes in SARS-CoV-2 and its impact on population infection characteristics
- PMID: 38348041
- PMCID: PMC10859446
- DOI: 10.3389/fimmu.2024.1341906
Bioinformatic analysis of defective viral genomes in SARS-CoV-2 and its impact on population infection characteristics
Abstract
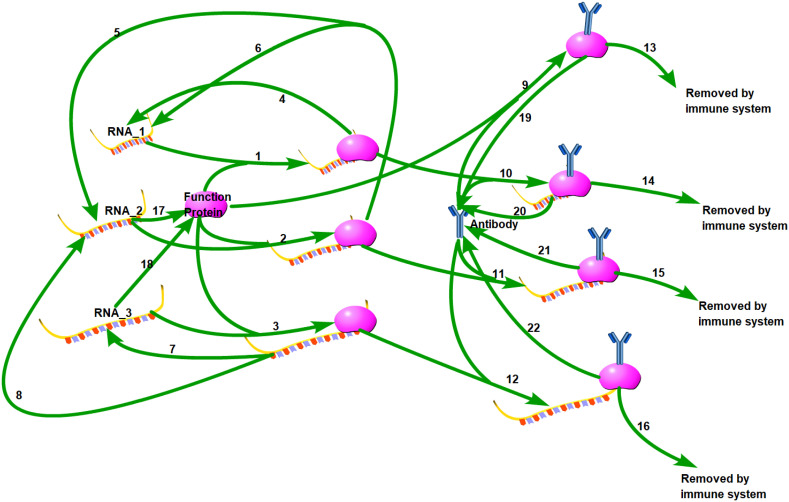
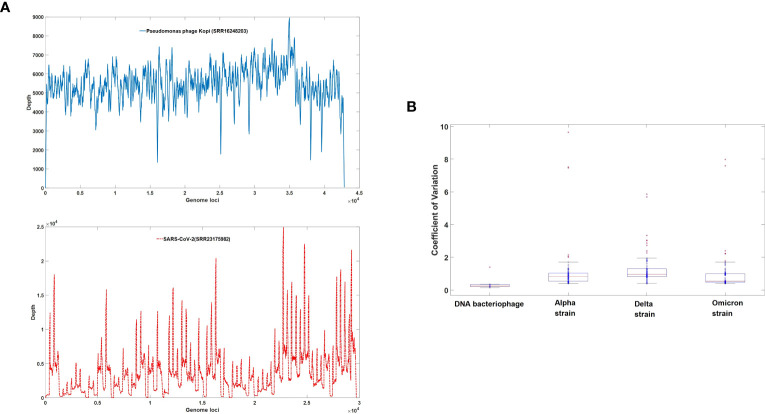
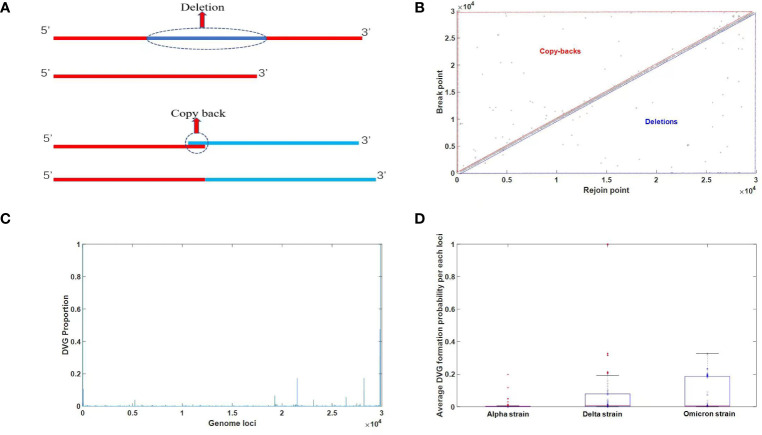
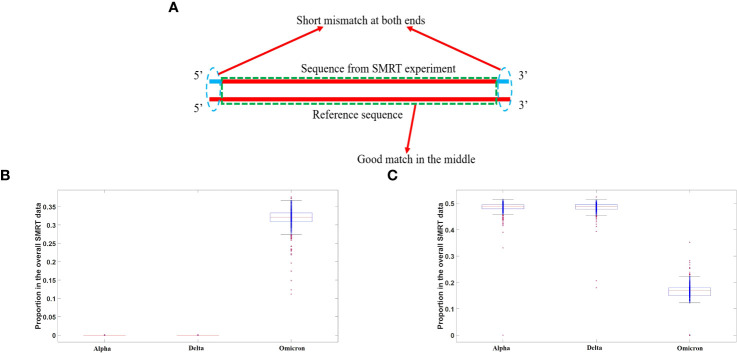
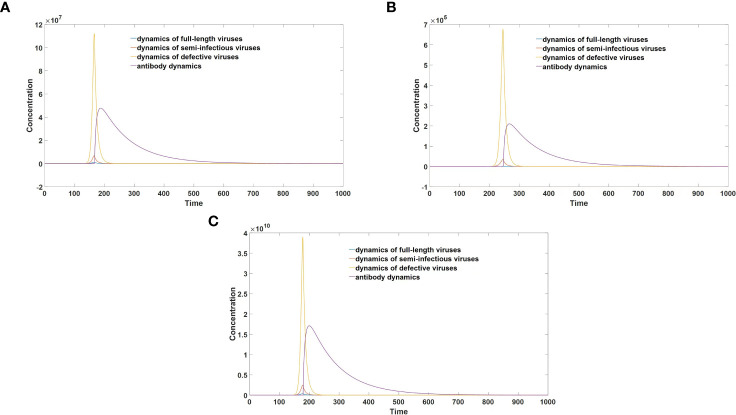
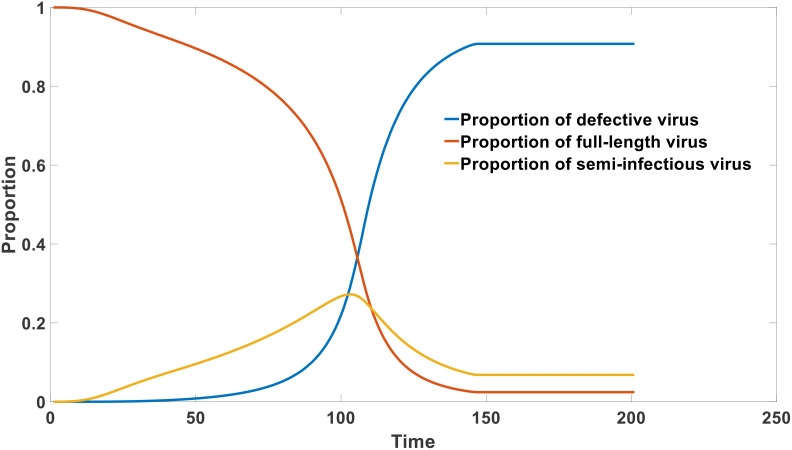
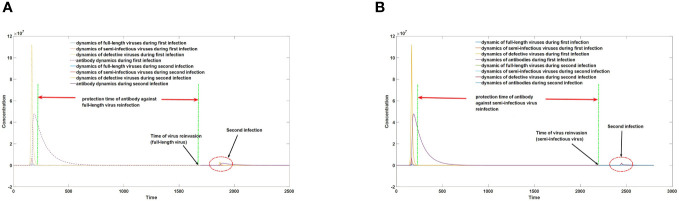
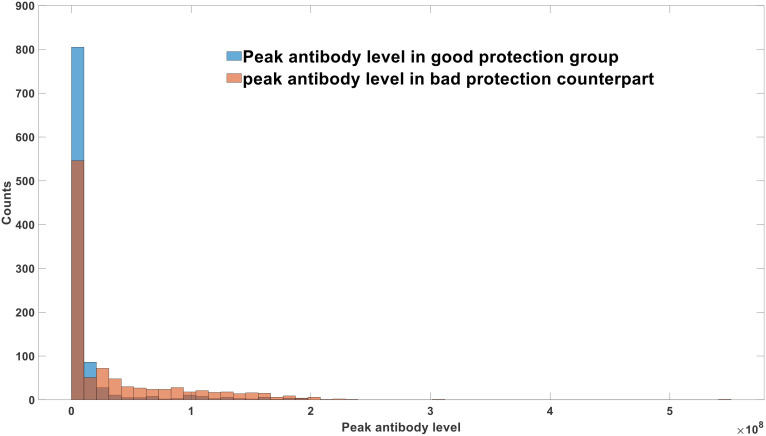
DVGs (Defective Viral Genomes) are prevalent in RNA virus infections. In this investigation, we conducted an analysis of high-throughput sequencing data and observed widespread presence of DVGs in SARS-CoV-2. Comparative analysis between SARS-CoV-2 and diverse DNA viruses revealed heightened susceptibility to damage and increased sequencing sample heterogeneity within the SARS-CoV-2 genome. Whole-genome sequencing depth variability analysis exhibited a higher coefficient of variation for SARS-CoV-2, while DVG analysis indicated a significant proportion of recombination sites, signifying notable genome heterogeneity and suggesting that a large proportion of assembled virus particles contain incomplete RNA sequences. Moreover, our investigation explored the sequencing depth and DVG content differences among various strains. Our findings revealed that as the virus evolves, there is a notable increase in the proportion of intact genomes within virus particles, as evidenced by third-generation sequencing data. Specifically, the proportion of intact genome in the Omicron strain surpassed that of the Delta and Alpha strains. This observation effectively elucidates the heightened infectiousness of the Omicron strain compared to the Delta and Alpha strains. We also postulate that this improvement in completeness stems from enhanced virus assembly capacity, as the Omicron strain can promptly facilitate the binding of RNA and capsid protein, thereby reducing the exposure time of vulnerable virus RNA in the host environment and significantly mitigating its degradation. Finally, employing mathematical modeling, we simulated the impact of DVG effects under varying environmental factors on infection characteristics and population evolution. Our findings provide an explanation for the close association between symptom severity and the extent of virus invasion, as well as the substantial disparity in population infection characteristics caused by the same strain under distinct environmental conditions. This study presents a novel approach for future virus research and vaccine development.
Keywords: SARS-CoV-2; defective viral genome; genome coverage; mathematical modeling; population infection characteristics; semi-infectious particle; virus evolution.
Copyright © 2024 Xu, Peng, Song, Zhang, Wei, Demongeot and Zeng.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
Comment on
-
Generation and Functional Analysis of Defective Viral Genomes during SARS-CoV-2 Infection.mBio. 2023 Jun 27;14(3):e0025023. doi: 10.1128/mbio.00250-23. Epub 2023 Apr 19. mBio. 2023. PMID: 37074178 Free PMC article.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous