

Distinct ontogenetic lineages dictate cDC2 heterogeneity
- PMID: 38351322
- PMCID: PMC10907303
- DOI: 10.1038/s41590-024-01745-9
Distinct ontogenetic lineages dictate cDC2 heterogeneity
Abstract
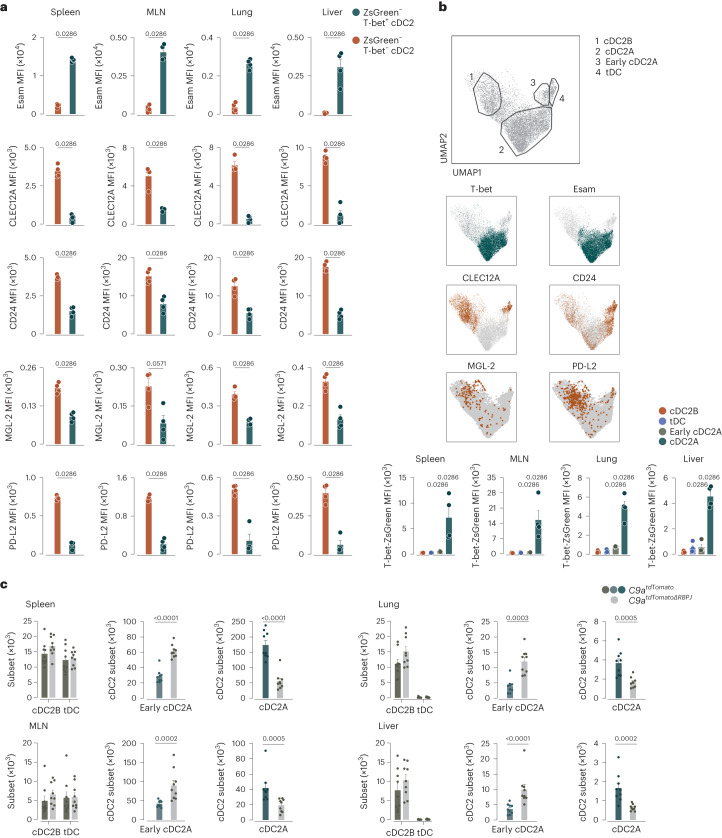
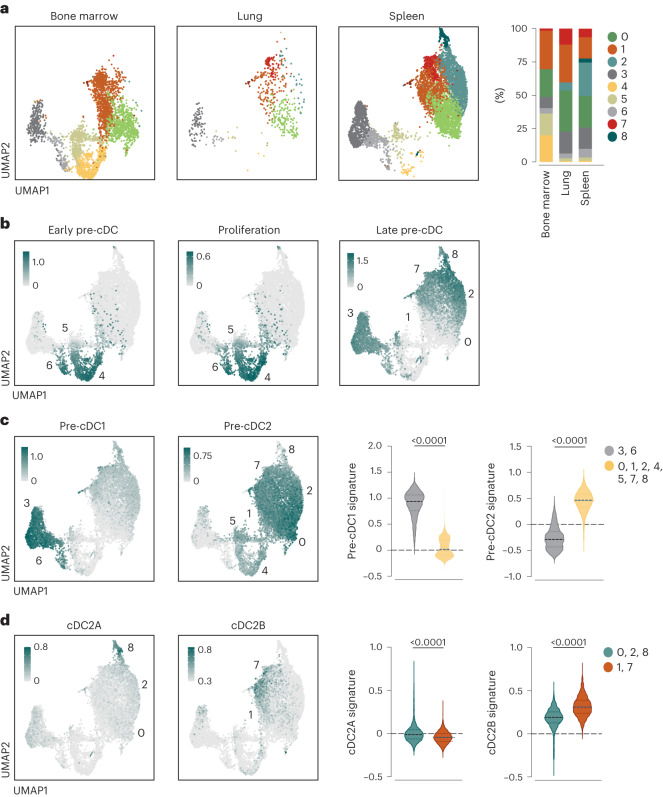
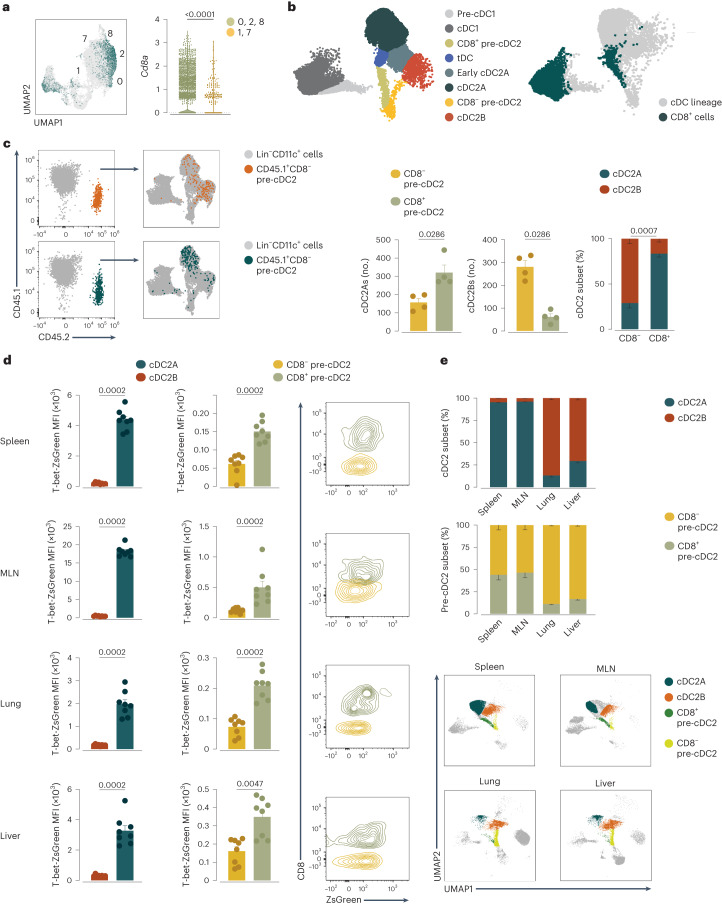
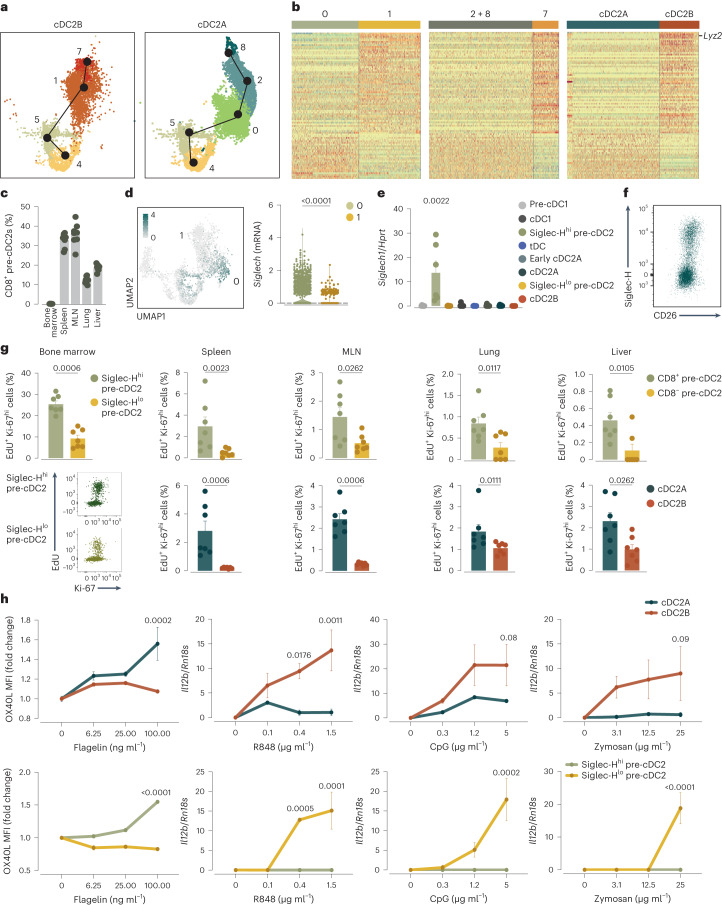
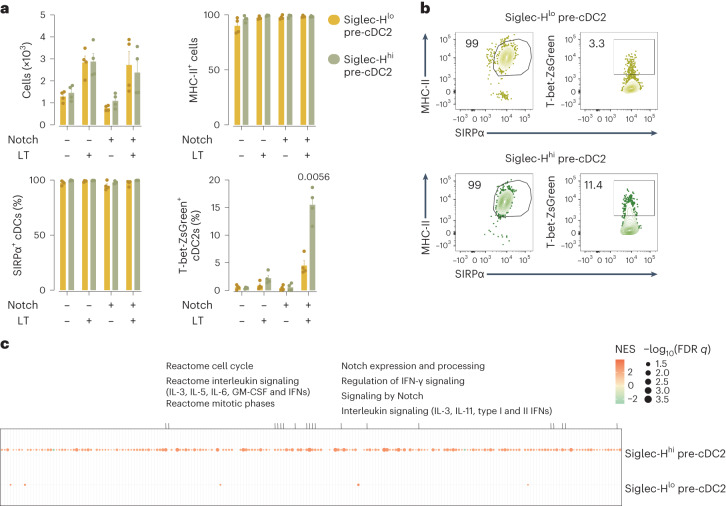
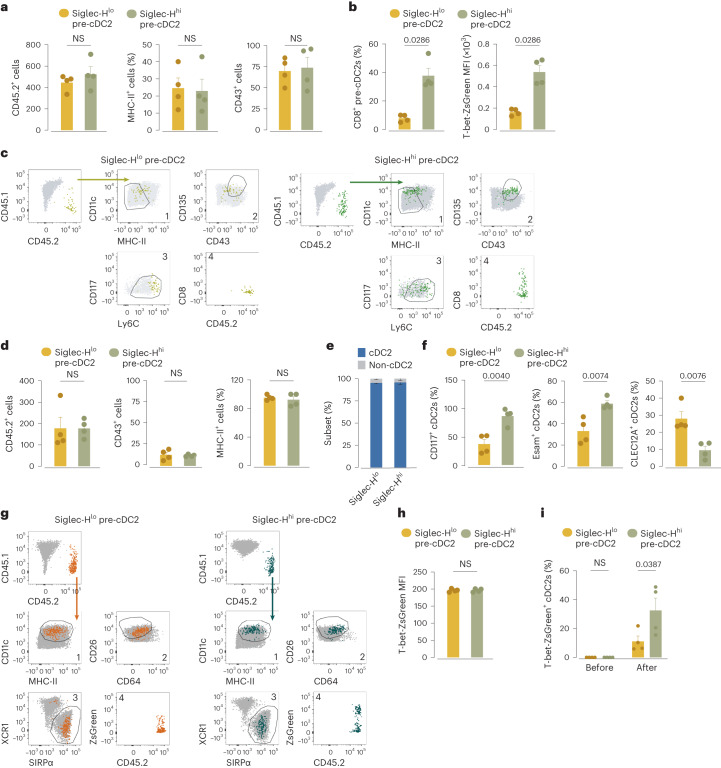
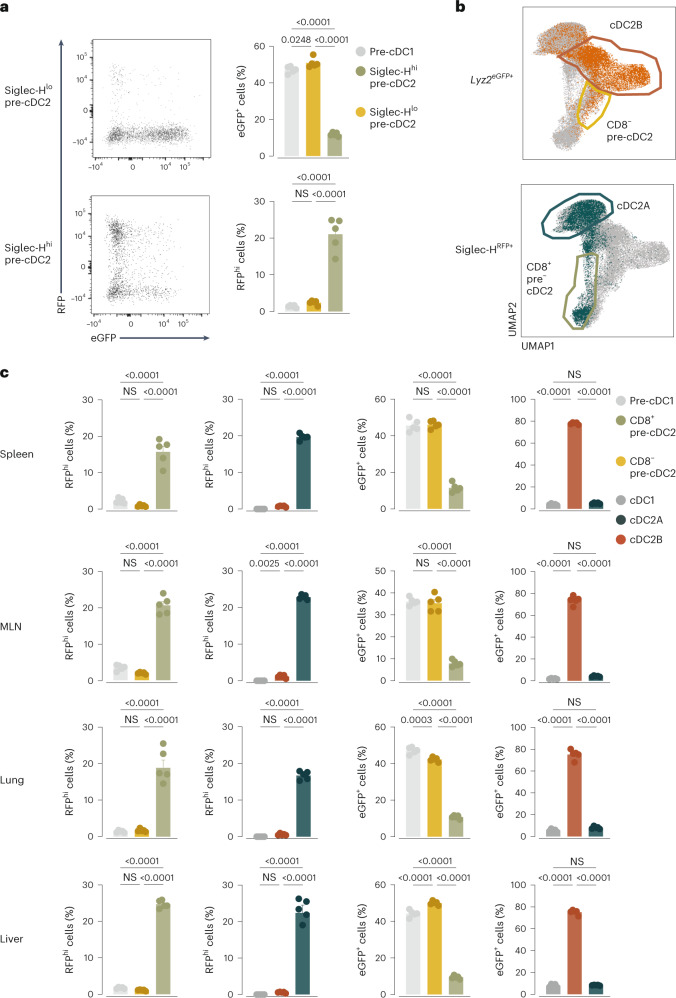
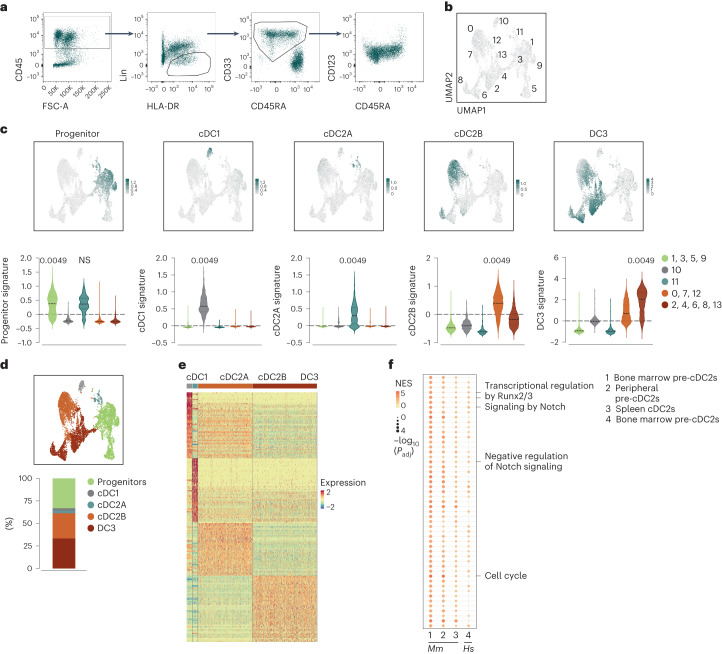
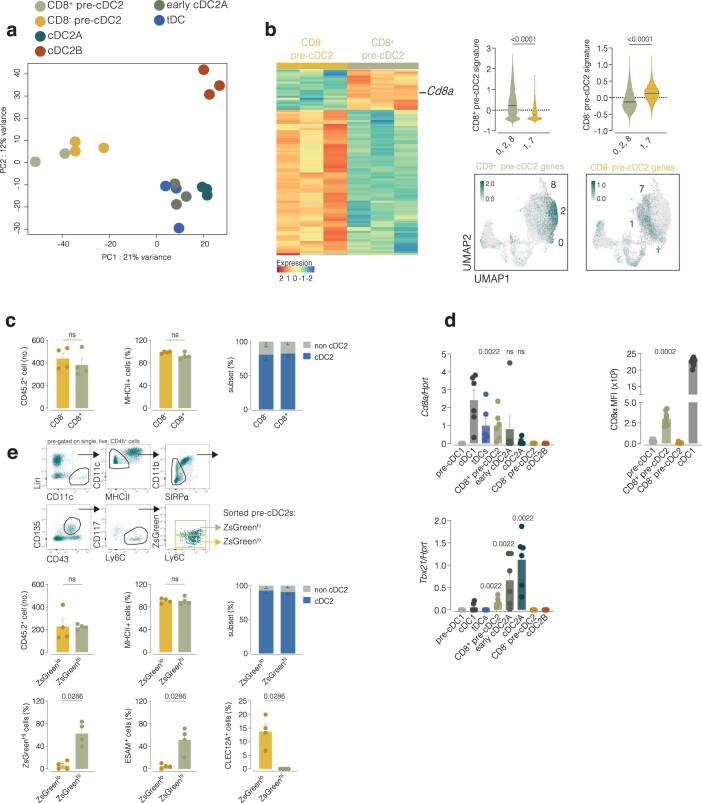
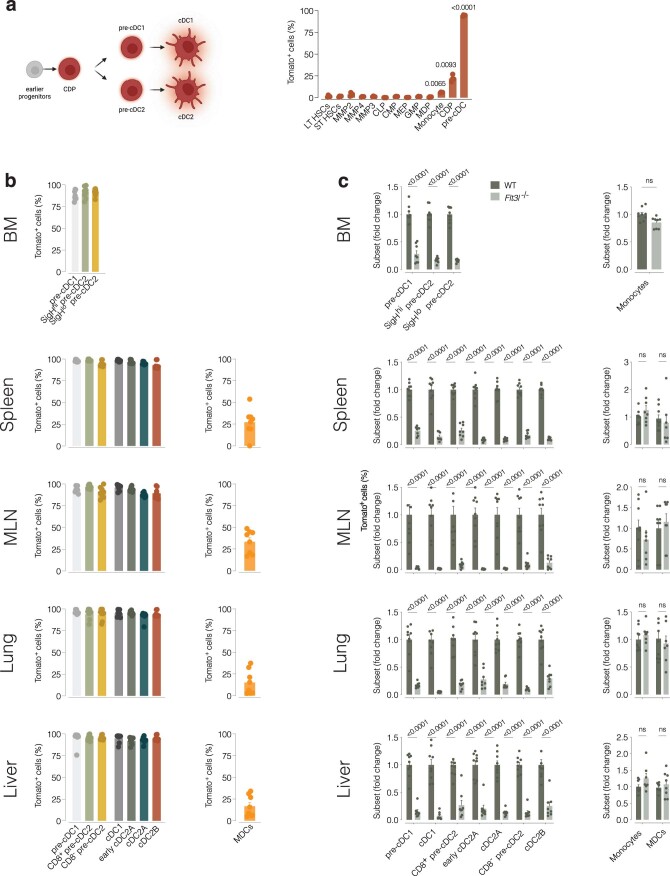
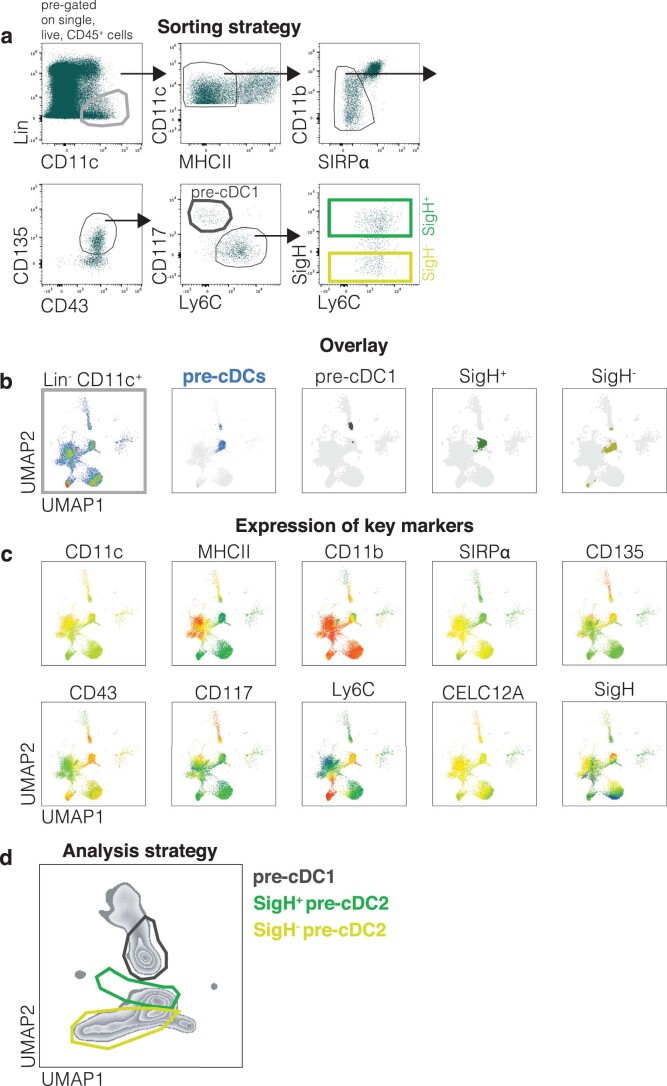
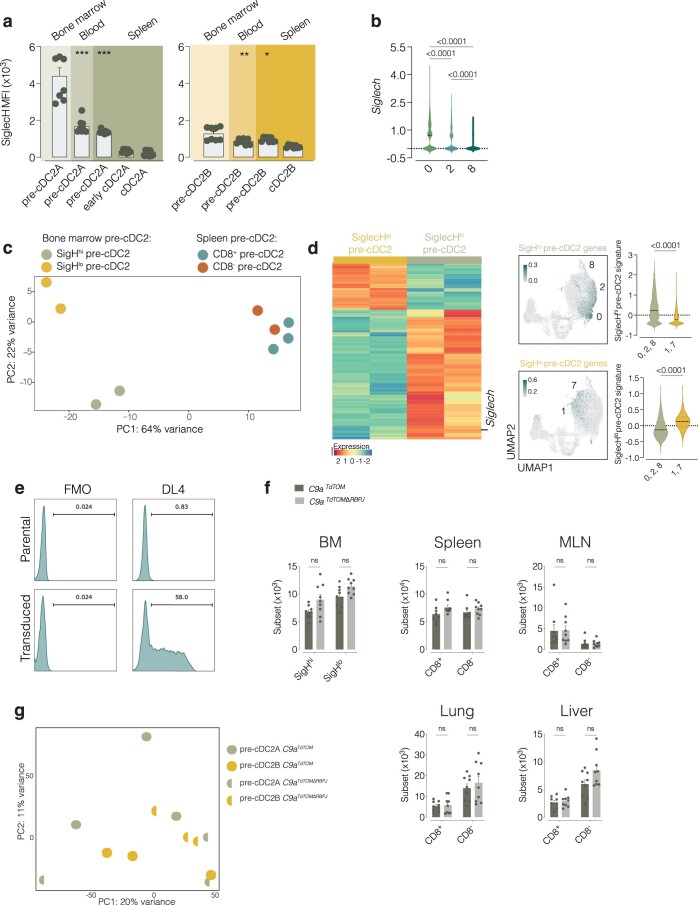
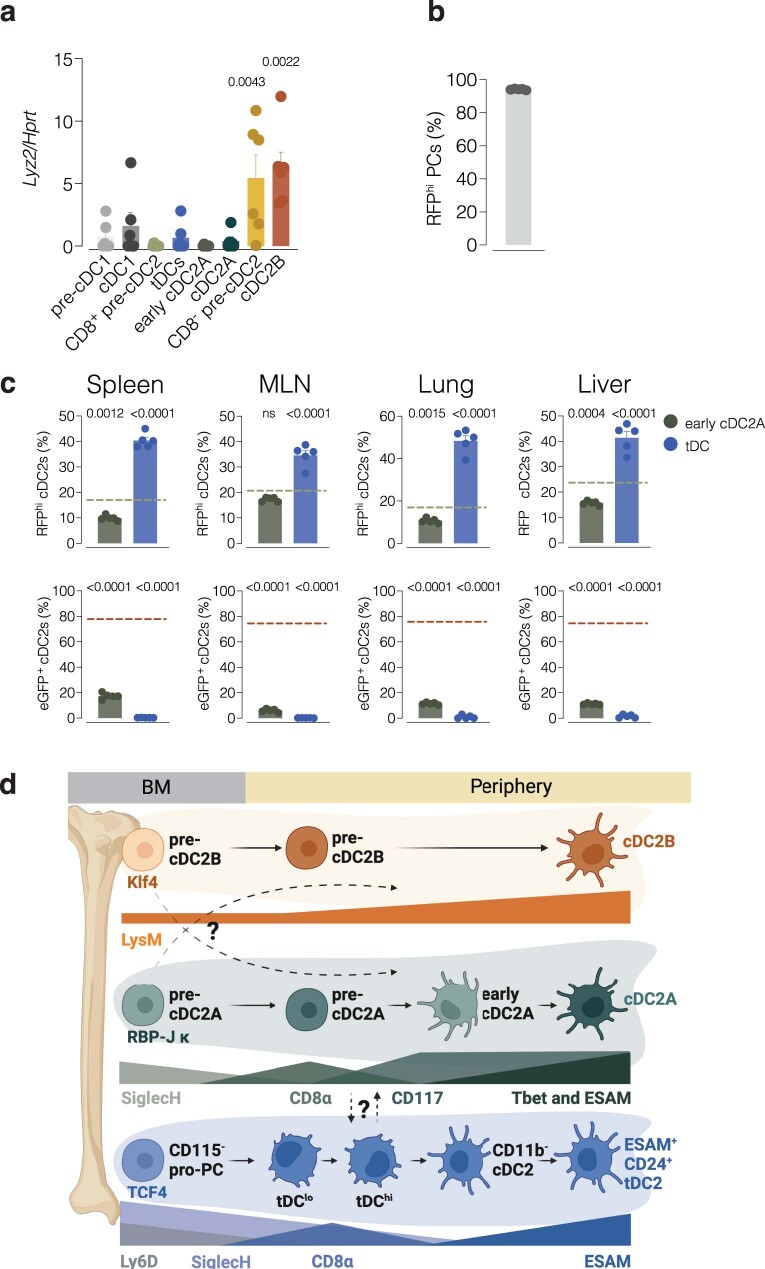
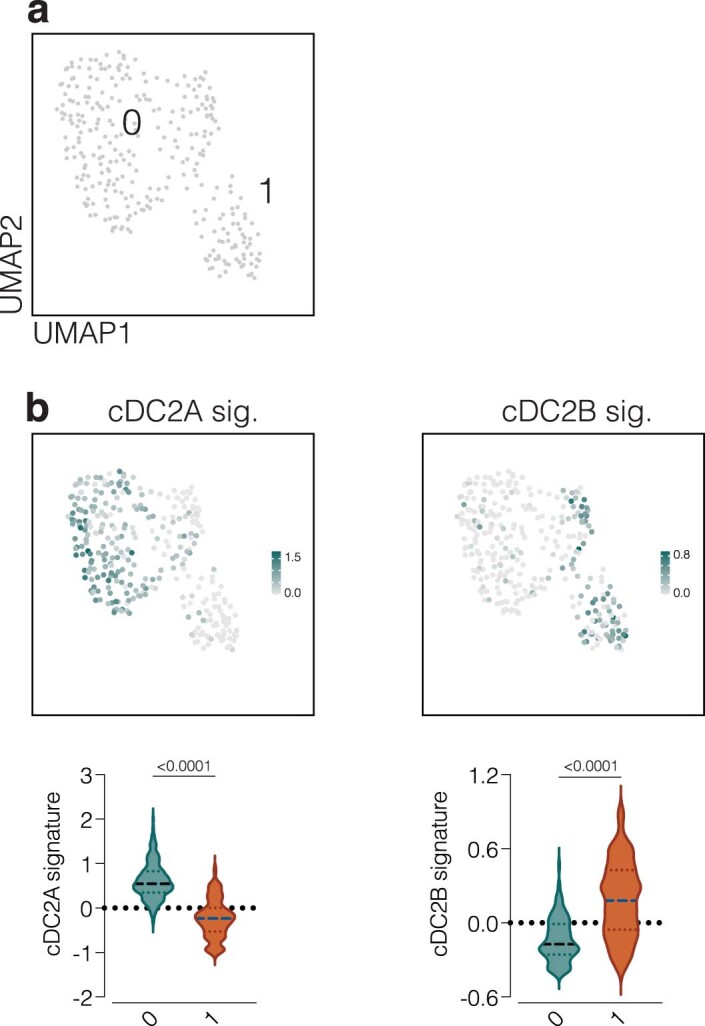
Conventional dendritic cells (cDCs) include functionally and phenotypically diverse populations, such as cDC1s and cDC2s. The latter population has been variously subdivided into Notch-dependent cDC2s, KLF4-dependent cDC2s, T-bet+ cDC2As and T-bet- cDC2Bs, but it is unclear how all these subtypes are interrelated and to what degree they represent cell states or cell subsets. All cDCs are derived from bone marrow progenitors called pre-cDCs, which circulate through the blood to colonize peripheral tissues. Here, we identified distinct mouse pre-cDC2 subsets biased to give rise to cDC2As or cDC2Bs. We showed that a Siglec-H+ pre-cDC2A population in the bone marrow preferentially gave rise to Siglec-H- CD8α+ pre-cDC2As in tissues, which differentiated into T-bet+ cDC2As. In contrast, a Siglec-H- fraction of pre-cDCs in the bone marrow and periphery mostly generated T-bet- cDC2Bs, a lineage marked by the expression of LysM. Our results showed that cDC2A versus cDC2B fate specification starts in the bone marrow and suggest that cDC2 subsets are ontogenetically determined lineages, rather than cell states imposed by the peripheral tissue environment.
© 2024. The Author(s).
Conflict of interest statement
C.R.S. has an additional appointment as visiting professor in the Faculty of Medicine at Imperial College London and holds honorary professorships at University College London and King’s College London. C.R.S. is a founder of Adendra Therapeutics and owns stock options in or is a paid consultant for Adendra Therapeutics, Bicara Therapeutics, Montis Biosciences and Bicycle Therapeutics, all unrelated to this work. The other authors declare no competing interests.
Figures
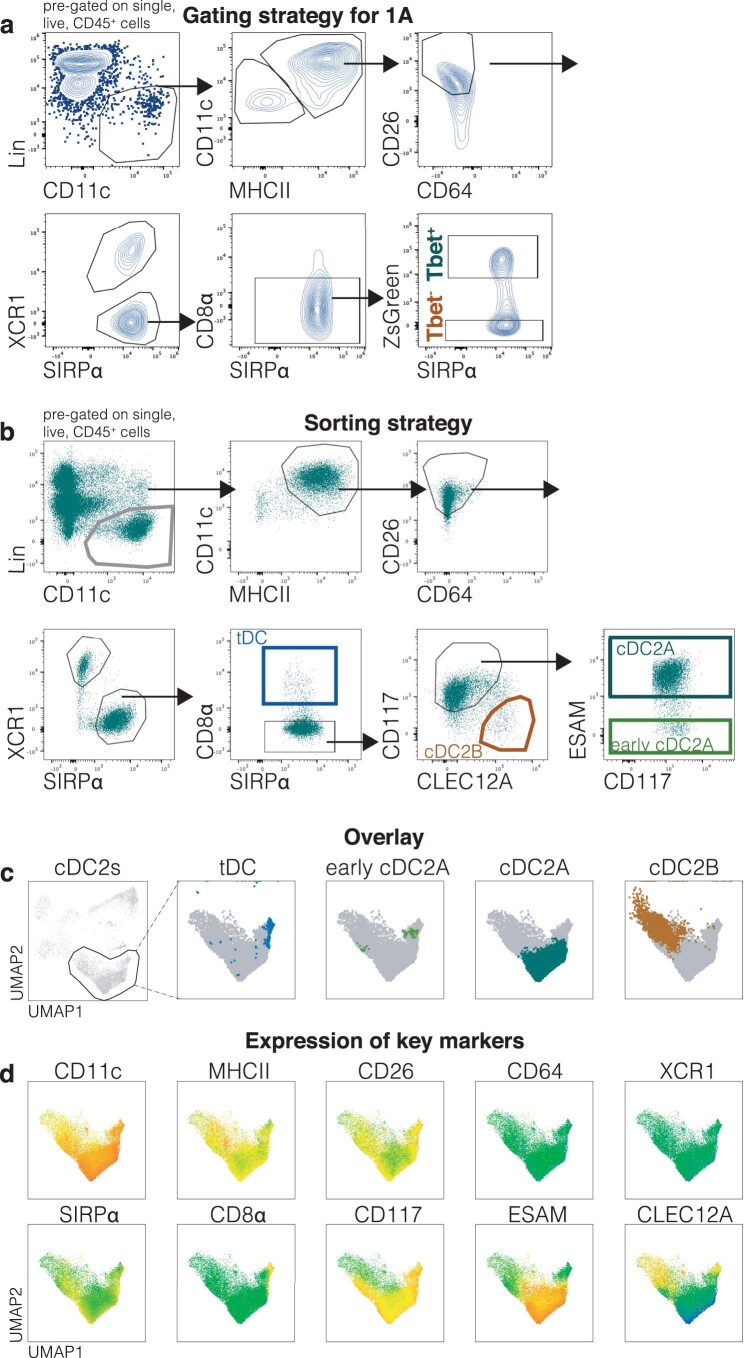
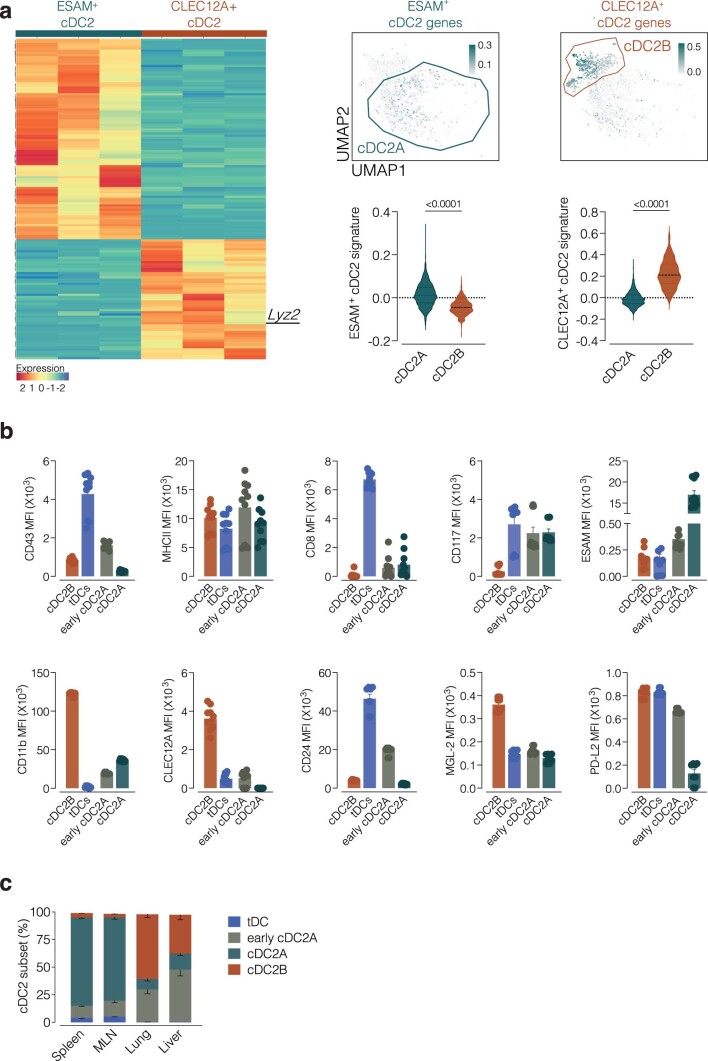
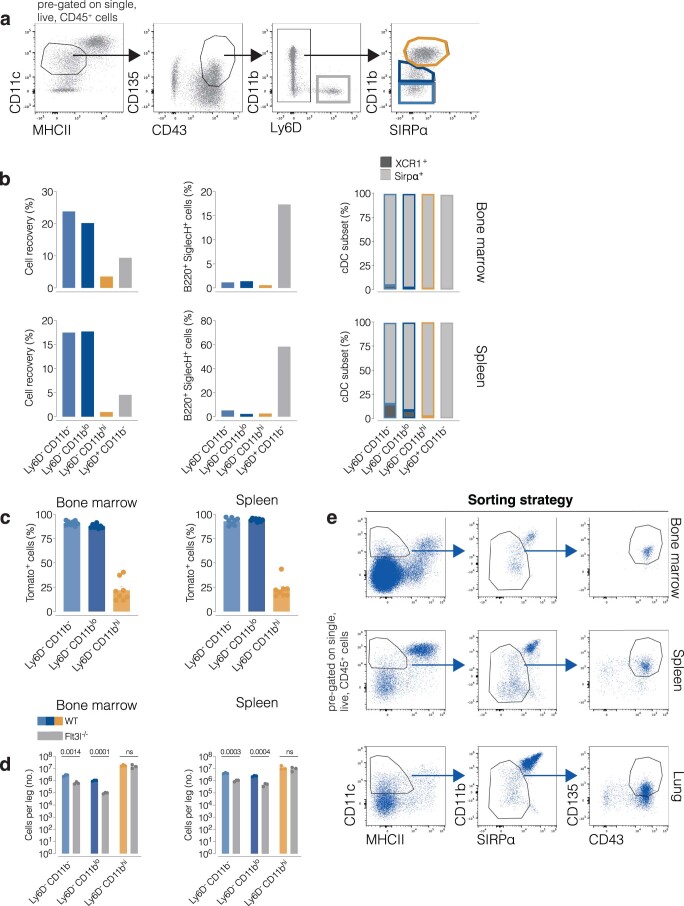
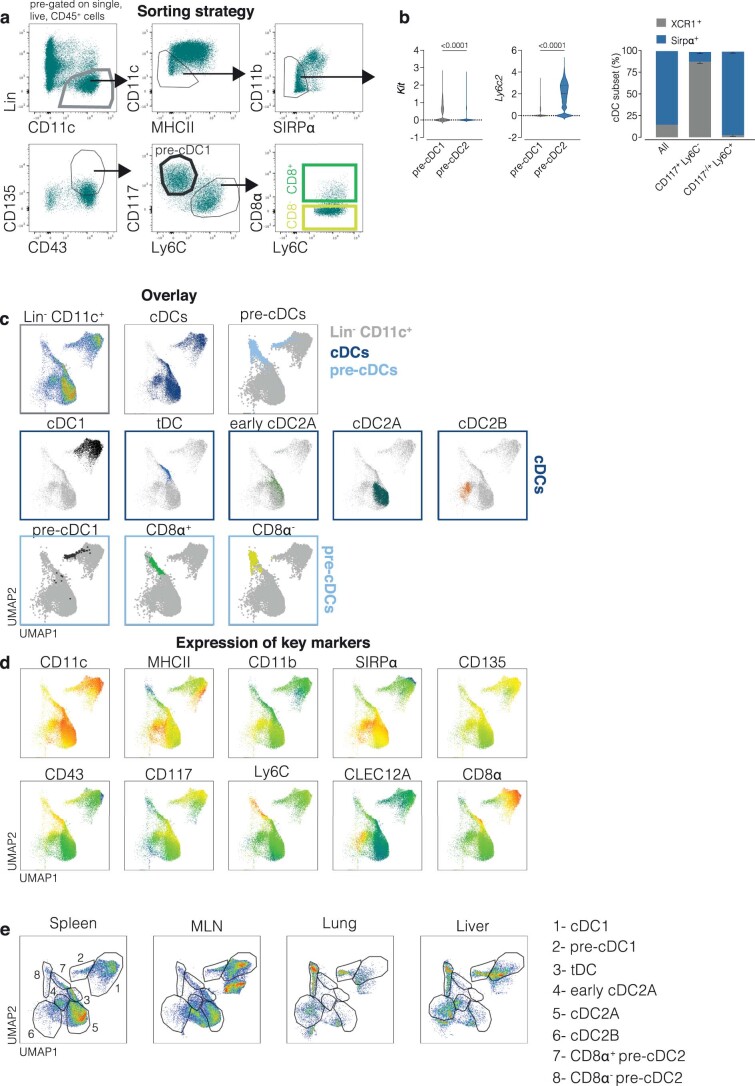
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
