

The role of programmed death receptor (PD-)1/PD-ligand (L)1 in periodontitis and cancer
- PMID: 38351432
- PMCID: PMC11579837
- DOI: 10.1111/prd.12548
The role of programmed death receptor (PD-)1/PD-ligand (L)1 in periodontitis and cancer
Abstract
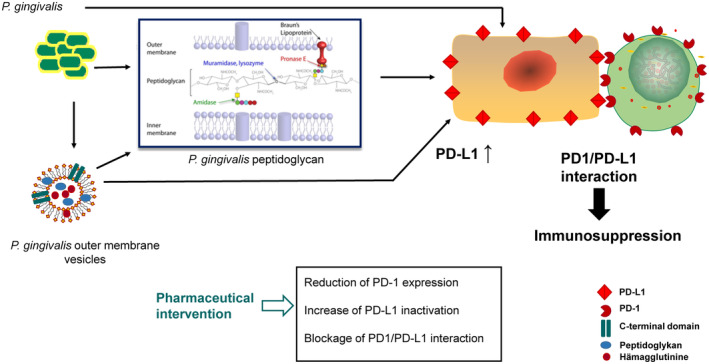
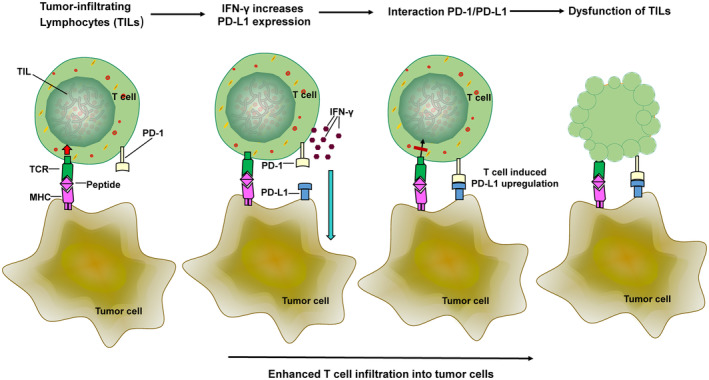
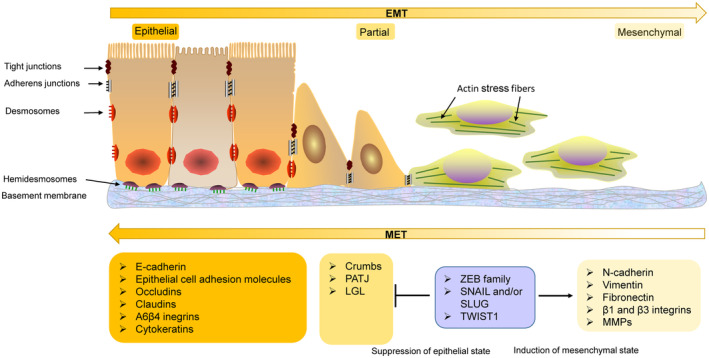
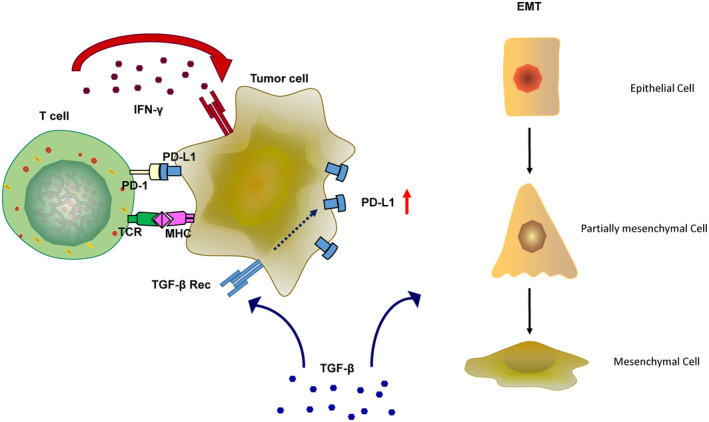
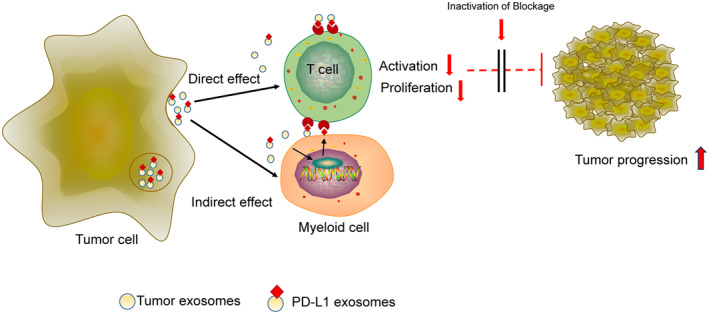
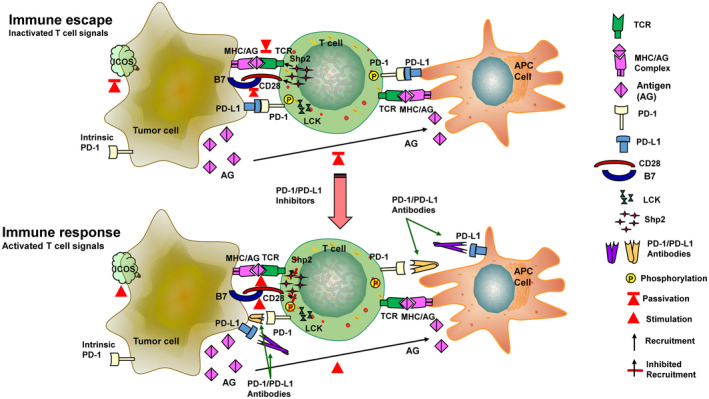
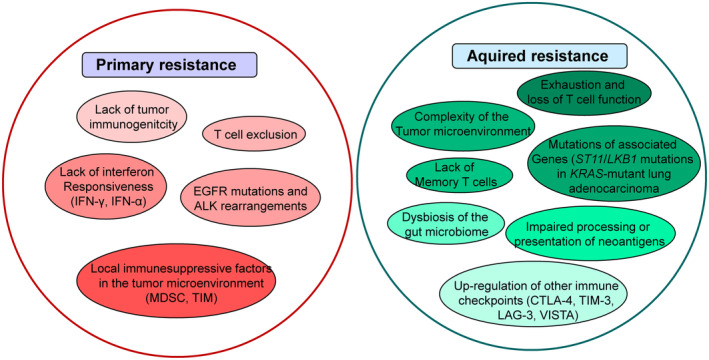
The programmed-death-ligand-1 (PD-L1) is an immune-modulating molecule that is constitutively expressed on various immune cells, different epithelial cells and a multitude of cancer cells. It is a costimulatory molecule that may impair T-cell mediated immune response. Ligation to the programmed-death-receptor (PD)-1, on activated T-cells and further triggering of the related signaling pathways can induce T-cells apoptosis or anergy. The upregulation of PD-L1 in various cancer types, including oral squamous cell carcinomas, was demonstrated and has been linked to immune escape of tumors and poor prognosis. A bidirectional relationship exists between the increased PD-L1 expression and periodontitis as well as the epithelial-mesenchymal transition (EMT), a process of interconversion of epithelial cells to mesenchymal cells that may induce immune escape of tumors. Interaction between exosomal PD-L1 and PD-1 on T-cells may cause immunosuppression by blocking the activation and proliferation of T-cells. The efficacy and importance of treatment with PD-1/PD-L1 checkpoint inhibitors and their prognostic influence on human cancers was demonstrated. Regarding PD-1/PD-L1 checkpoint inhibitors, resistances exist or may develop, basing on various factors. Further investigations of the underlying mechanisms will help to overcome the therapeutic limitations that result from resistances and to develop new strategies for the treatment of cancer.
Keywords: EMT; PD‐L1; cancer; exosomes; immune checkpoint; immune escape; periodontitis.
© 2024 The Authors. Periodontology 2000 published by John Wiley & Sons Ltd.
Conflict of interest statement
The authors declare that they have no conflicts of interest.
Figures
References
-
- Darveau RP. Periodontitis: a polymicrobial disruption of host homeostasis. Nat Rev Microbiol. 2010;8:481‐490. - PubMed
-
- Larsen T, Fiehn NE. Dental biofilm infections—an update. APMIS. 2017;125:376‐384. - PubMed
-
- Picolos DK, Lerche‐Sehm J, Abron A, Fine JB, Papapanou PN. Infection patterns in chronic and aggressive periodontitis. J Clin Periodontol. 2005;32:1055‐1061. - PubMed
-
- Dong H, Strome SE, Salomao DR, et al. Tumor‐associated B7‐H1 promotes T‐cell apoptosis: a potential mechanism of immune evasion. Nat Med. 2002;8:793‐800. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
