

Comprehensive overview of the role of mitochondrial dysfunction in the pathogenesis of acute kidney ischemia-reperfusion injury: a narrative review
- PMID: 38351610
- PMCID: PMC11074843
- DOI: 10.12701/jyms.2023.01347
Comprehensive overview of the role of mitochondrial dysfunction in the pathogenesis of acute kidney ischemia-reperfusion injury: a narrative review
Abstract
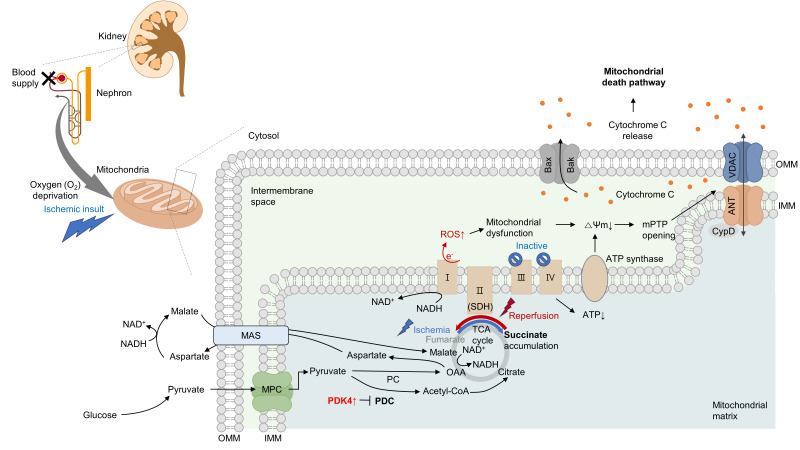
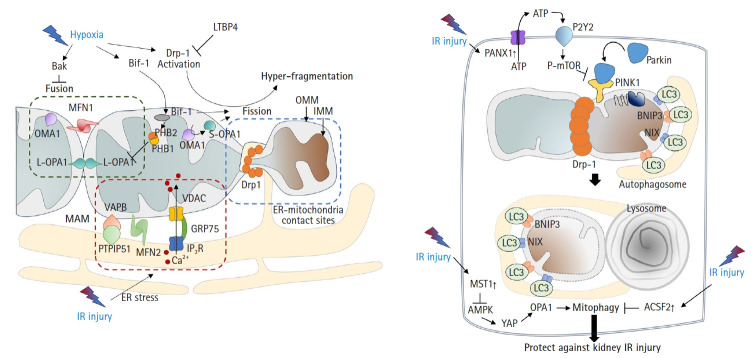
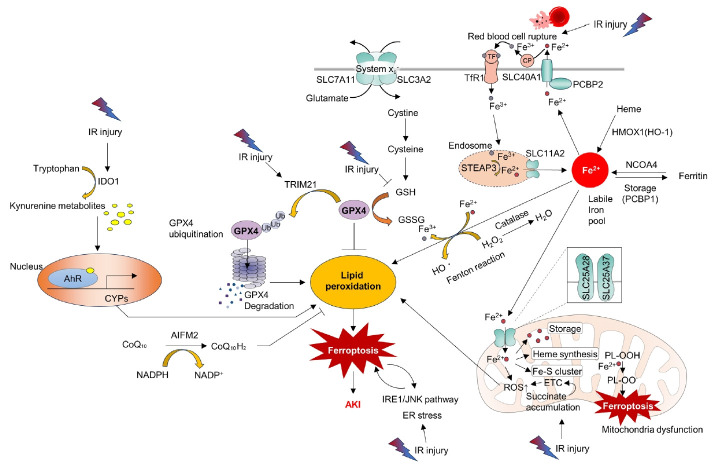
Acute kidney ischemia-reperfusion (IR) injury is a life-threatening condition that predisposes individuals to chronic kidney disease. Since the kidney is one of the most energy-demanding organs in the human body and mitochondria are the powerhouse of cells, mitochondrial dysfunction plays a central role in the pathogenesis of IR-induced acute kidney injury. Mitochondrial dysfunction causes a reduction in adenosine triphosphate production, loss of mitochondrial dynamics (represented by persistent fragmentation), and impaired mitophagy. Furthermore, the pathological accumulation of succinate resulting from fumarate reduction under oxygen deprivation (ischemia) in the reverse flux of the Krebs cycle can eventually lead to a burst of reactive oxygen species driven by reverse electron transfer during the reperfusion phase. Accumulating evidence indicates that improving mitochondrial function, biogenesis, and dynamics, and normalizing metabolic reprogramming within the mitochondria have the potential to preserve kidney function during IR injury and prevent progression to chronic kidney disease. In this review, we summarize recent advances in understanding the detrimental role of metabolic reprogramming and mitochondrial dysfunction in IR injury and explore potential therapeutic strategies for treating kidney IR injury.
Keywords: Acute kidney injury; Ferroptosis; Mitochondria; Mitochondrial dynamics; Mitophagy; Reactive oxygen species.
Conflict of interest statement
No potential conflict of interest relevant to this article was reported.
Figures
Similar articles
-
Inhibition of pyruvate dehydrogenase kinase 4 ameliorates kidney ischemia-reperfusion injury by reducing succinate accumulation during ischemia and preserving mitochondrial function during reperfusion.Kidney Int. 2023 Oct;104(4):724-739. doi: 10.1016/j.kint.2023.06.022. Epub 2023 Jul 1. Kidney Int. 2023. PMID: 37399974
-
Mitochondria ROS and mitophagy in acute kidney injury.Autophagy. 2023 Feb;19(2):401-414. doi: 10.1080/15548627.2022.2084862. Epub 2022 Jun 9. Autophagy. 2023. PMID: 35678504 Free PMC article.
-
High-fat diet-induced mitochondrial dysfunction is associated with loss of protection from ischemic preconditioning in renal ischemia reperfusion.Pflugers Arch. 2023 May;475(5):637-653. doi: 10.1007/s00424-023-02799-8. Epub 2023 Mar 3. Pflugers Arch. 2023. PMID: 36867229
-
Mitophagy and mitochondrial integrity in cardiac ischemia-reperfusion injury.Biochim Biophys Acta Mol Basis Dis. 2019 Sep 1;1865(9):2293-2302. doi: 10.1016/j.bbadis.2019.05.007. Epub 2019 May 14. Biochim Biophys Acta Mol Basis Dis. 2019. PMID: 31100337 Review.
-
Mitochondrial Quality Control in Cerebral Ischemia-Reperfusion Injury.Mol Neurobiol. 2021 Oct;58(10):5253-5271. doi: 10.1007/s12035-021-02494-8. Epub 2021 Jul 18. Mol Neurobiol. 2021. PMID: 34275087 Review.
Cited by
-
Spatiotemporal transcriptomic analysis during cold ischemic injury to the murine kidney reveals compartment-specific changes.bioRxiv [Preprint]. 2025 May 28:2025.05.25.654911. doi: 10.1101/2025.05.25.654911. bioRxiv. 2025. PMID: 40501673 Free PMC article. Preprint.
-
Hyperoside mitigates amphotericin B-induced nephrotoxicity in HK-2 cells via bioenergetic and oxidative stress modulation.BMC Pharmacol Toxicol. 2025 Aug 12;26(1):149. doi: 10.1186/s40360-025-00985-1. BMC Pharmacol Toxicol. 2025. PMID: 40797341 Free PMC article.
References
-
- Khwaja A. KDIGO clinical practice guidelines for acute kidney injury. Nephron Clin Pract. 2012;120:c179–84. - PubMed
-
- Rewa O, Bagshaw SM. Acute kidney injury-epidemiology, outcomes and economics. Nat Rev Nephrol. 2014;10:193–207. - PubMed
-
- Sabouny R, Shutt TE. The role of mitochondrial dynamics in mtDNA maintenance. J Cell Sci. 2021;134:jcs258944. - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
