

This is a preprint.
Reference-free Structural Variant Detection in Microbiomes via Long-read Coassembly Graphs
- PMID: 38352454
- PMCID: PMC10862772
- DOI: 10.1101/2024.01.25.577285
Reference-free Structural Variant Detection in Microbiomes via Long-read Coassembly Graphs
Update in
-
Reference-free structural variant detection in microbiomes via long-read co-assembly graphs.Bioinformatics. 2024 Jun 28;40(Suppl 1):i58-i67. doi: 10.1093/bioinformatics/btae224. Bioinformatics. 2024. PMID: 38940156 Free PMC article.
Abstract
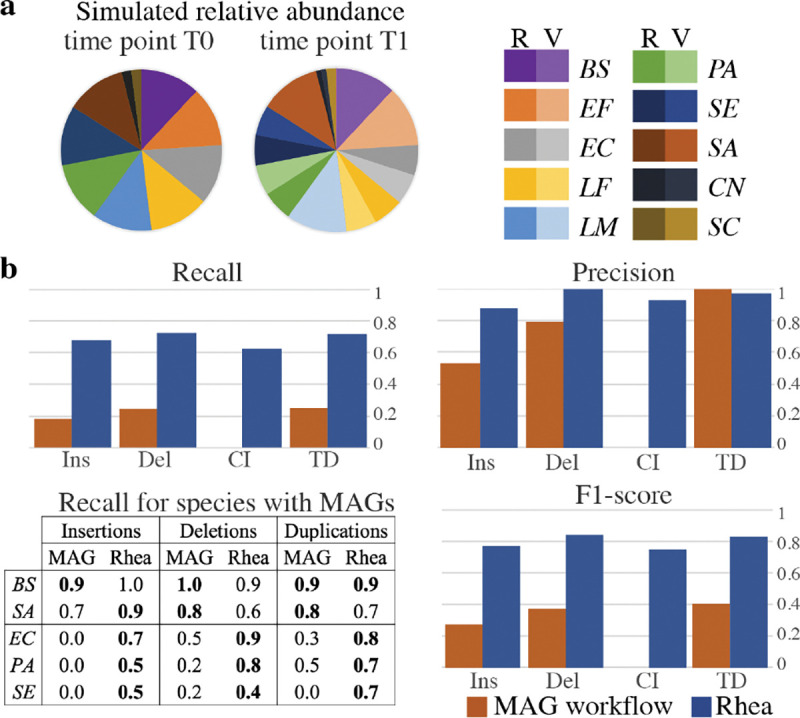
Bacterial genome dynamics are vital for understanding the mechanisms underlying microbial adaptation, growth, and their broader impact on host phenotype. Structural variants (SVs), genomic alterations of 10 base pairs or more, play a pivotal role in driving evolutionary processes and maintaining genomic heterogeneity within bacterial populations. While SV detection in isolate genomes is relatively straightforward, metagenomes present broader challenges due to absence of clear reference genomes and presence of mixed strains. In response, our proposed method rhea, forgoes reference genomes and metagenome-assembled genomes (MAGs) by encompassing a single metagenome coassembly graph constructed from all samples in a series. The log fold change in graph coverage between subsequent samples is then calculated to call SVs that are thriving or declining throughout the series. We show rhea to outperform existing methods for SV and horizontal gene transfer (HGT) detection in two simulated mock metagenomes, which is particularly noticeable as the simulated reads diverge from reference genomes and an increase in strain diversity is incorporated. We additionally demonstrate use cases for rhea on series metagenomic data of environmental and fermented food microbiomes to detect specific sequence alterations between subsequent time and temperature samples, suggesting host advantage. Our innovative approach leverages raw read patterns rather than references or MAGs to include all sequencing reads in analysis, and thus provide versatility in studying SVs across diverse and poorly characterized microbial communities for more comprehensive insights into microbial genome dynamics.
Keywords: gene transfer; long-read sequencing; metagenome; microbiome; structural variants.
Conflict of interest statement
Competing interests No competing interest is declared.
Figures



References
-
- Balaji A., Liu Y., Nute M.G., Hu B., D. Kappell A., S. Lesassier D., D. Godbold G., Ternus K., Treangen T.: SeqScreen-Nano: A computational platform for streaming, in-field characterization of microbial pathogens. In: Proceedings of the 14th ACM International Conference on Bioinformatics, Computational Biology, and Health Informatics. pp. 1–10. BCB ‘23, Association for Computing Machinery, New York, NY, USA (Oct 2023). 10.1145/3584371.3612960 - DOI
-
- Bhaya D., Grossman A.R., Steunou A.S., Khuri N., Cohan F.M., Hamamura N., Melendrez M.C., Bateson M.M., Ward D.M., Heidelberg J.F.: Population level functional diversity in a microbial community revealed by comparative genomic and metagenomic analyses. The ISME journal 1(8), 703–713 (Dec 2007). 10.1038/ismej.2007.46 - DOI - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous