

This is a preprint.
Non-CG DNA methylation and MeCP2 stabilize repeated tuning of long genes that distinguish closely related neuron types
- PMID: 38352532
- PMCID: PMC10862856
- DOI: 10.1101/2024.01.30.577861
Non-CG DNA methylation and MeCP2 stabilize repeated tuning of long genes that distinguish closely related neuron types
Update in
-
MeCP2 and non-CG DNA methylation stabilize the expression of long genes that distinguish closely related neuron types.Nat Neurosci. 2025 Jun;28(6):1185-1198. doi: 10.1038/s41593-025-01947-w. Epub 2025 May 12. Nat Neurosci. 2025. PMID: 40355611
Abstract
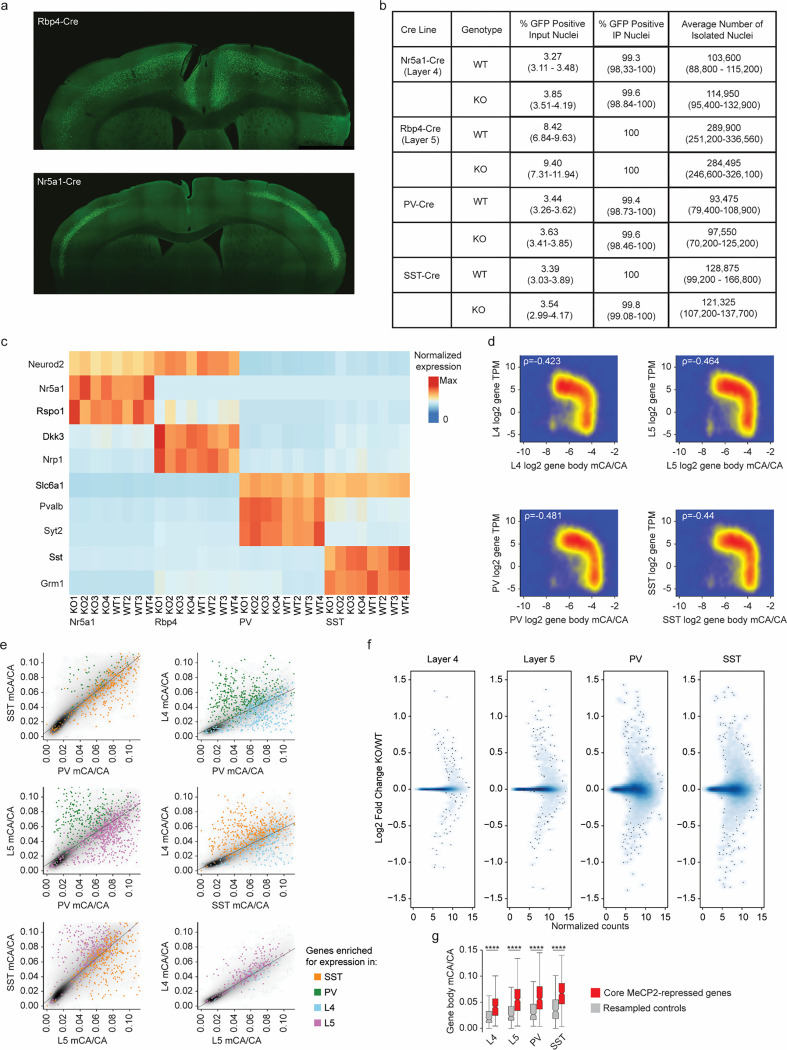
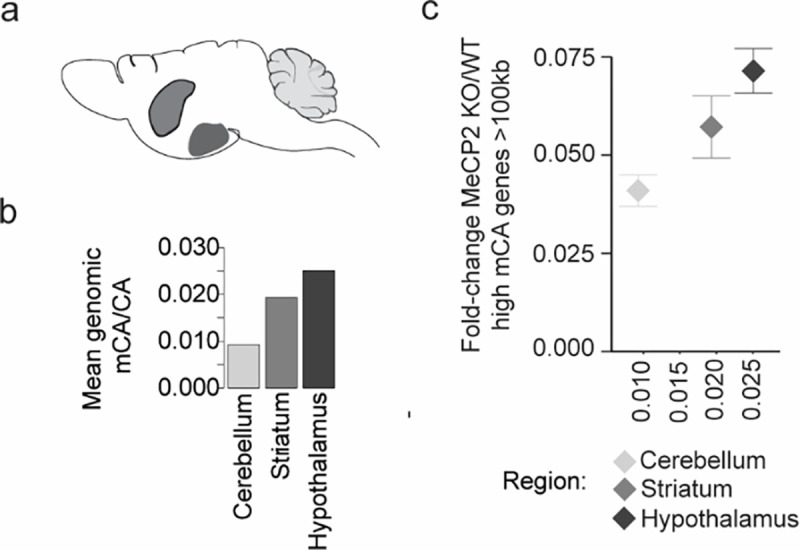
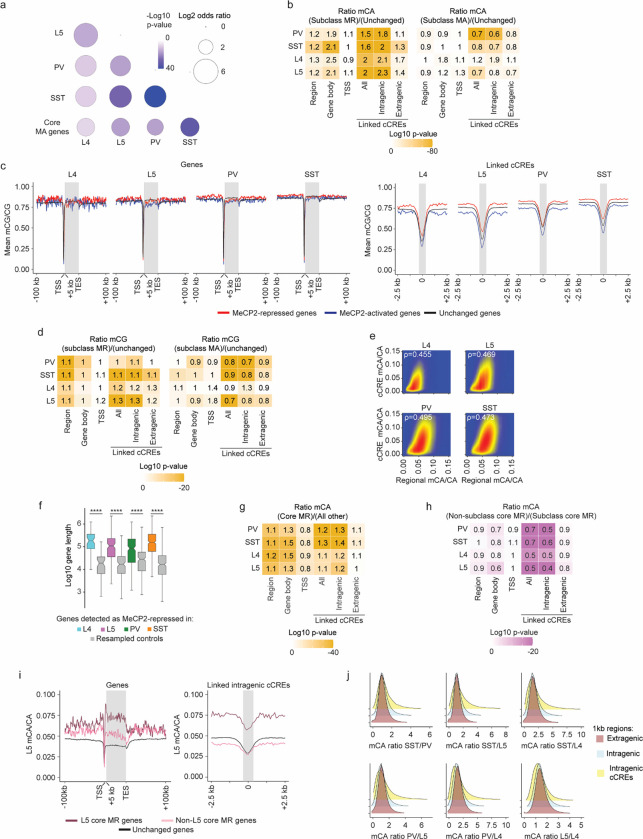
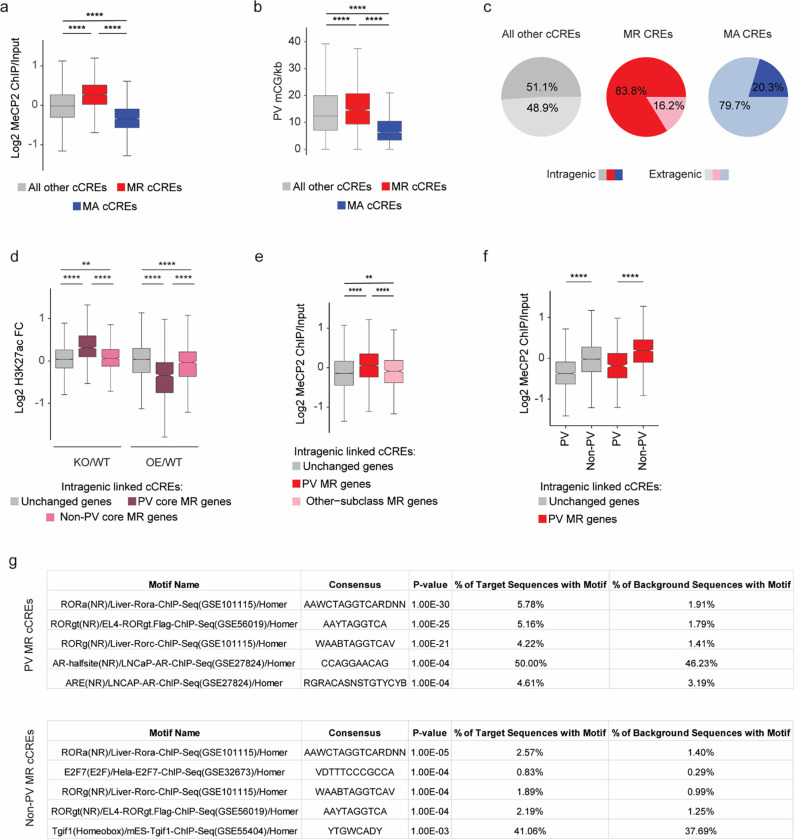
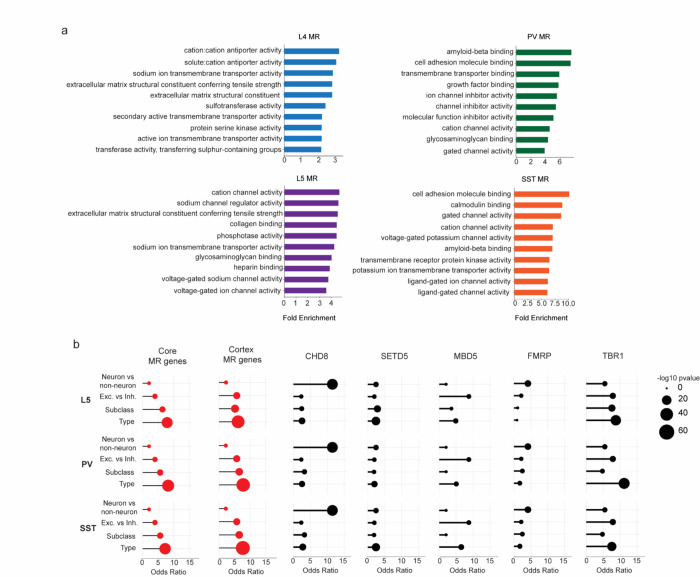
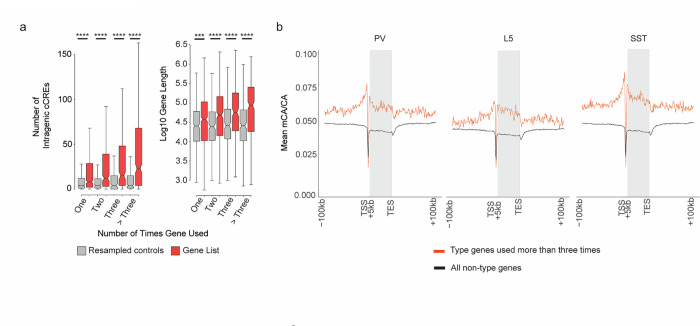
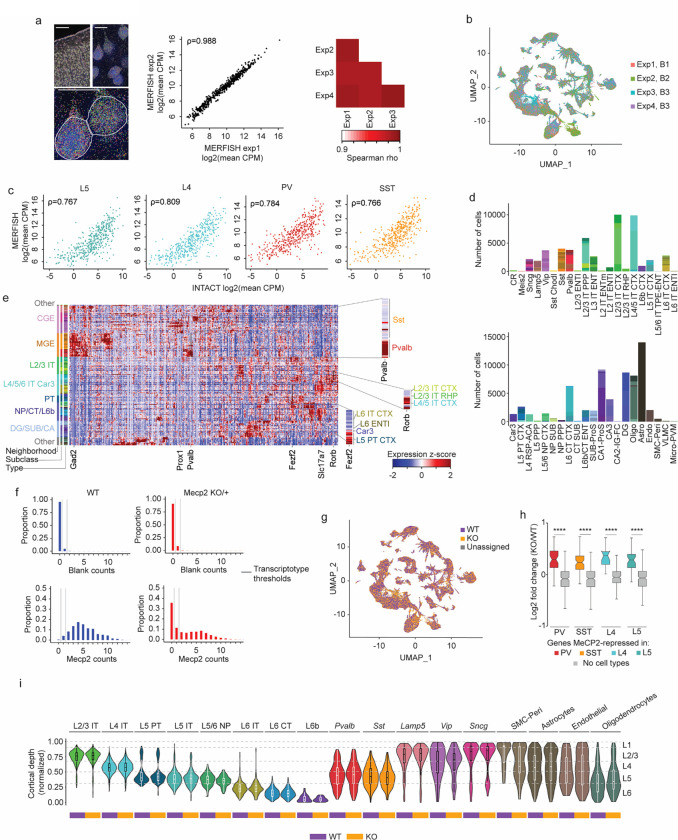
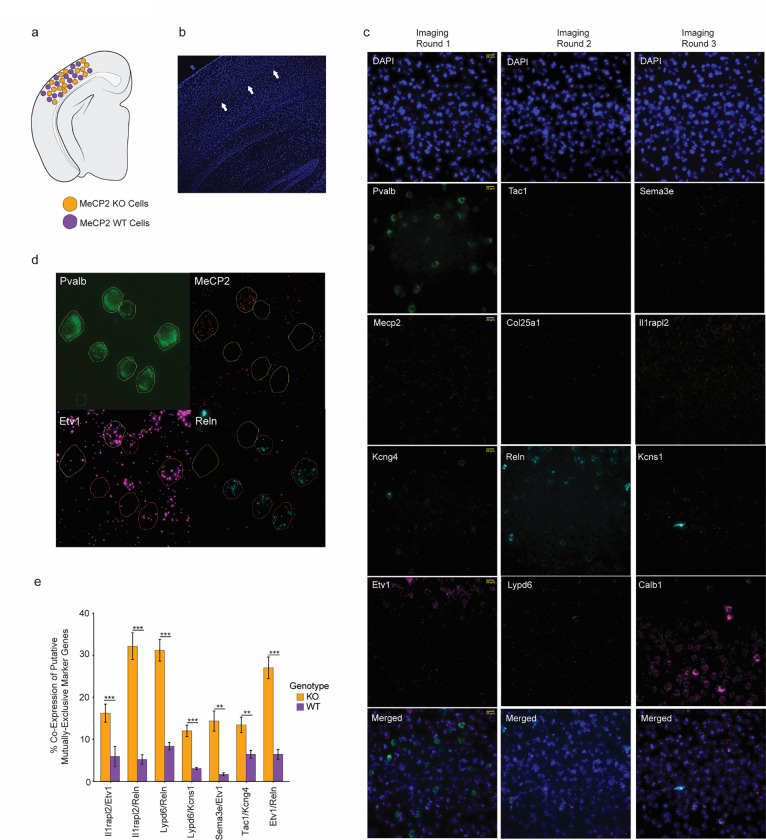
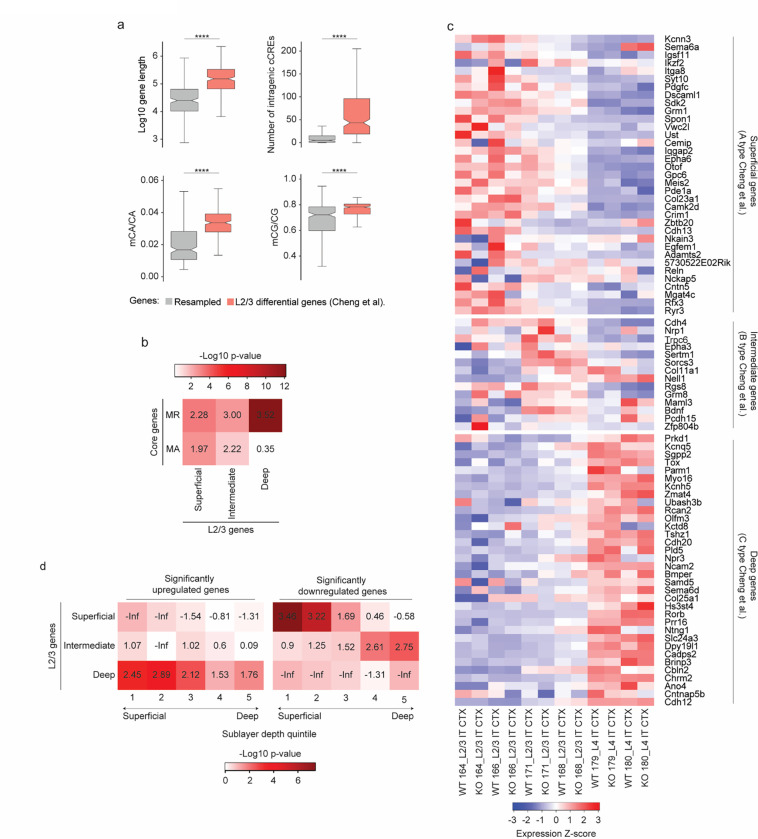
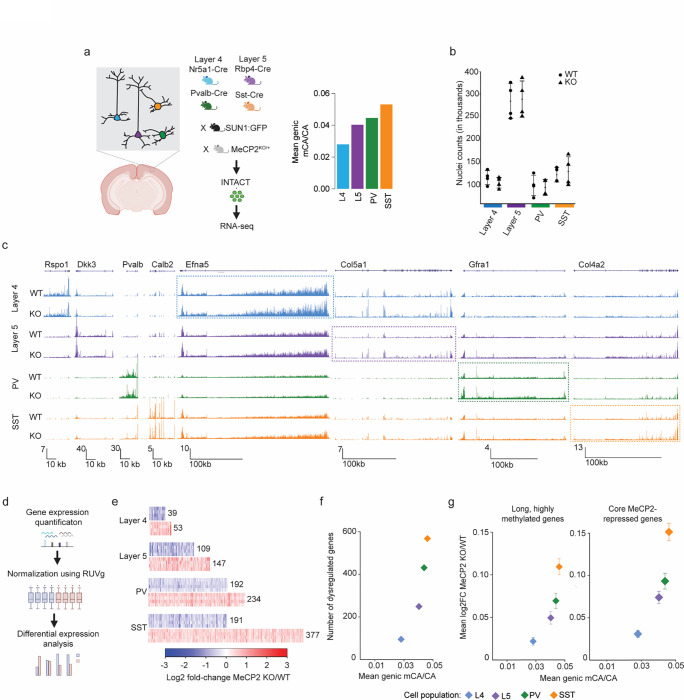
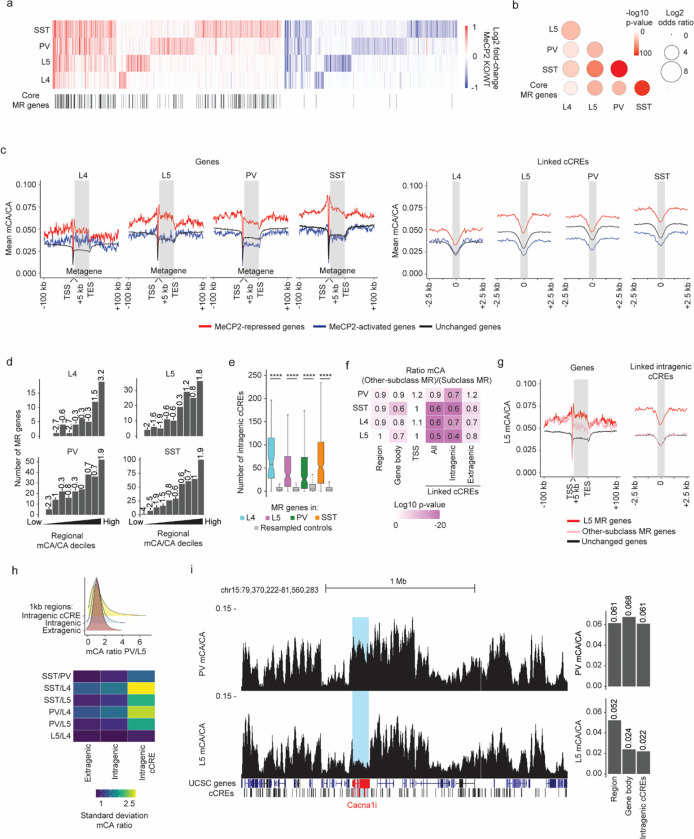
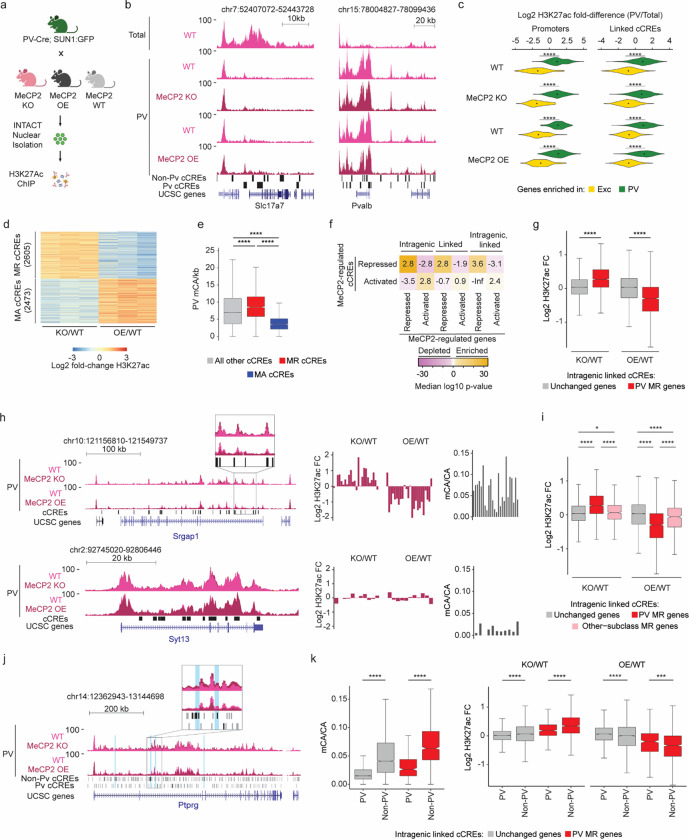
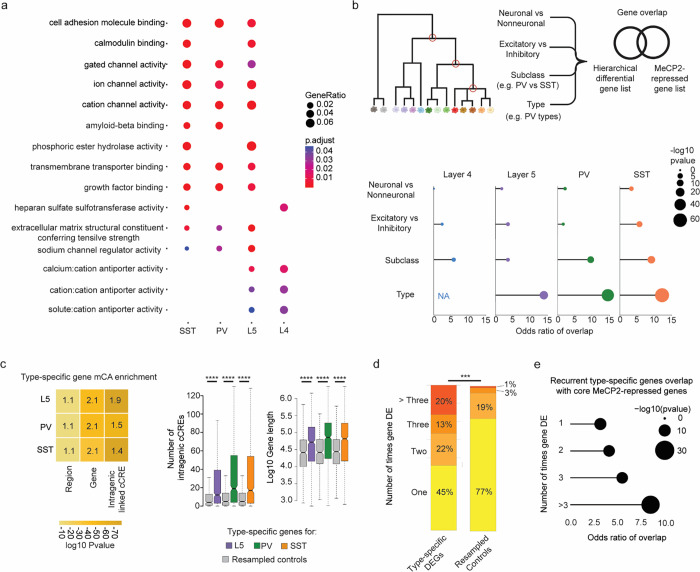
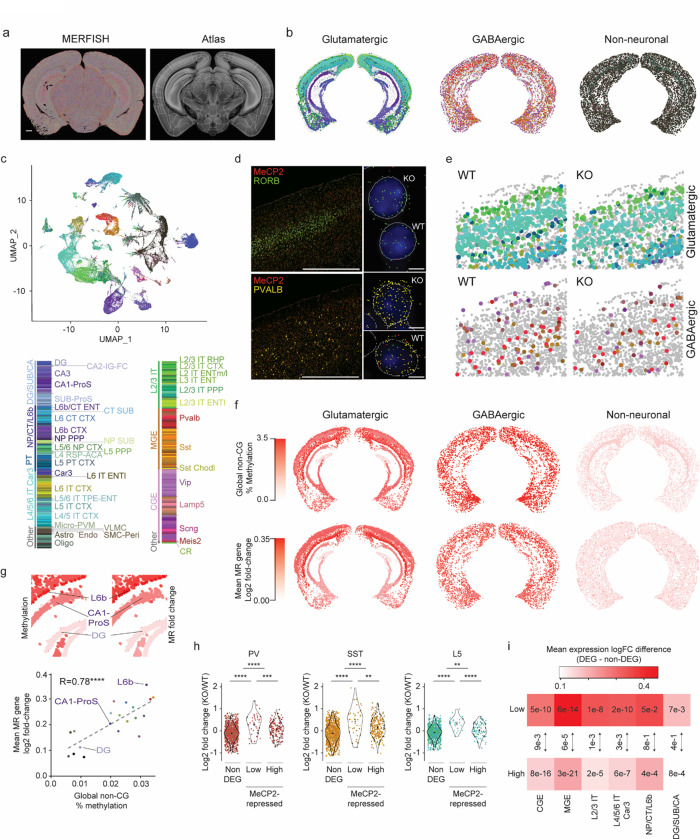
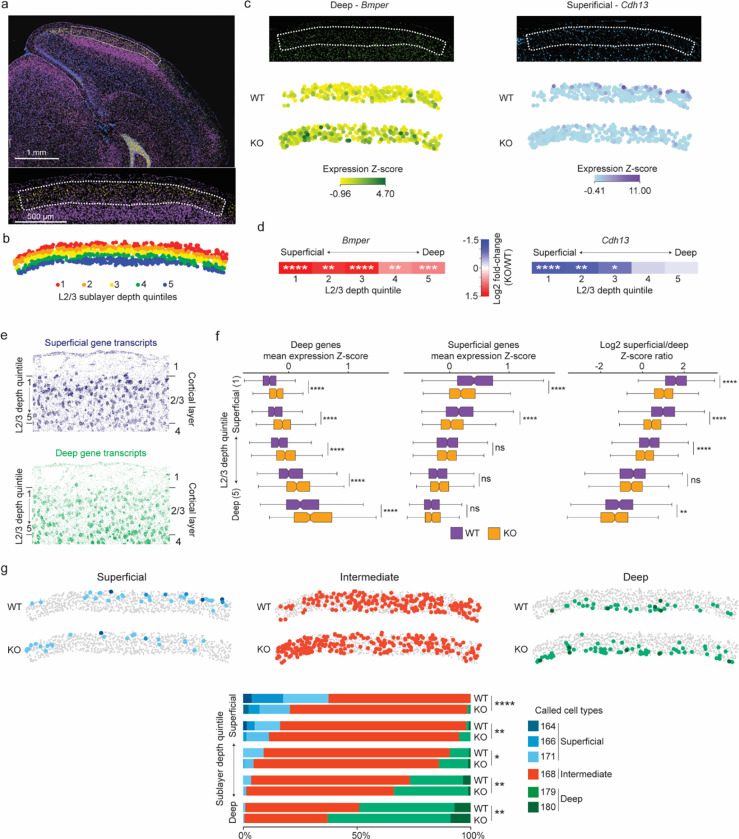
The extraordinary diversity of neuron types in the mammalian brain is delineated at the highest resolution by subtle gene expression differences that may require specialized molecular mechanisms to be maintained. Neurons uniquely express the longest genes in the genome and utilize neuron-enriched non-CG DNA methylation (mCA) together with the Rett syndrome protein, MeCP2, to control gene expression, but the function of these unique gene structures and machinery in regulating finely resolved neuron type-specific gene programs has not been explored. Here, we employ epigenomic and spatial transcriptomic analyses to discover a major role for mCA and MeCP2 in maintaining neuron type-specific gene programs at the finest scale of cellular resolution. We uncover differential susceptibility to MeCP2 loss in neuronal populations depending on global mCA levels and dissect methylation patterns and intragenic enhancer repression that drive overlapping and distinct gene regulation between neuron types. Strikingly, we show that mCA and MeCP2 regulate genes that are repeatedly tuned to differentiate neuron types at the highest cellular resolution, including spatially resolved, vision-dependent gene programs in the visual cortex. These repeatedly tuned genes display genomic characteristics, including long length, numerous intragenic enhancers, and enrichment for mCA, that predispose them to regulation by MeCP2. Thus, long gene regulation by the MeCP2 pathway maintains differential gene expression between closely-related neurons to facilitate the exceptional cellular diversity in the complex mammalian brain.
Figures
References
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources