

Construction of disulfidptosis-based immune response prediction model with artificial intelligence and validation of the pivotal grouping oncogene c-MET in regulating T cell exhaustion
- PMID: 38352883
- PMCID: PMC10862485
- DOI: 10.3389/fimmu.2024.1258475
Construction of disulfidptosis-based immune response prediction model with artificial intelligence and validation of the pivotal grouping oncogene c-MET in regulating T cell exhaustion
Abstract
Background: Given the lack of research on disulfidptosis, our study aimed to dissect its role in pan-cancer and explore the crosstalk between disulfidptosis and cancer immunity.
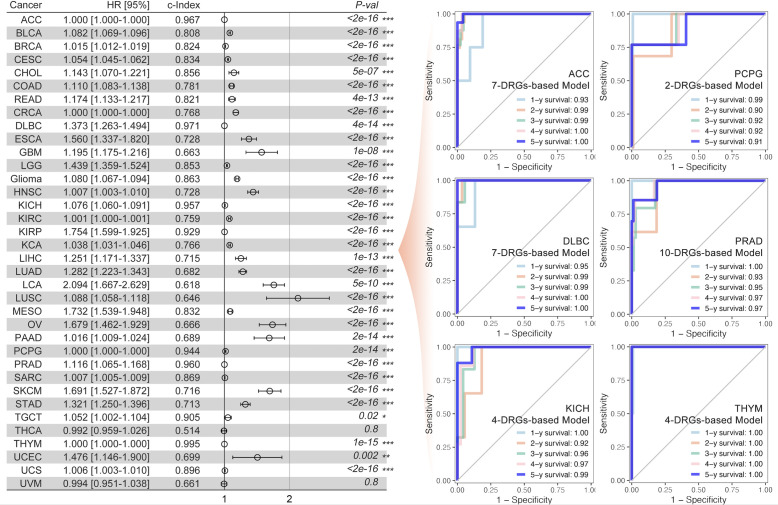
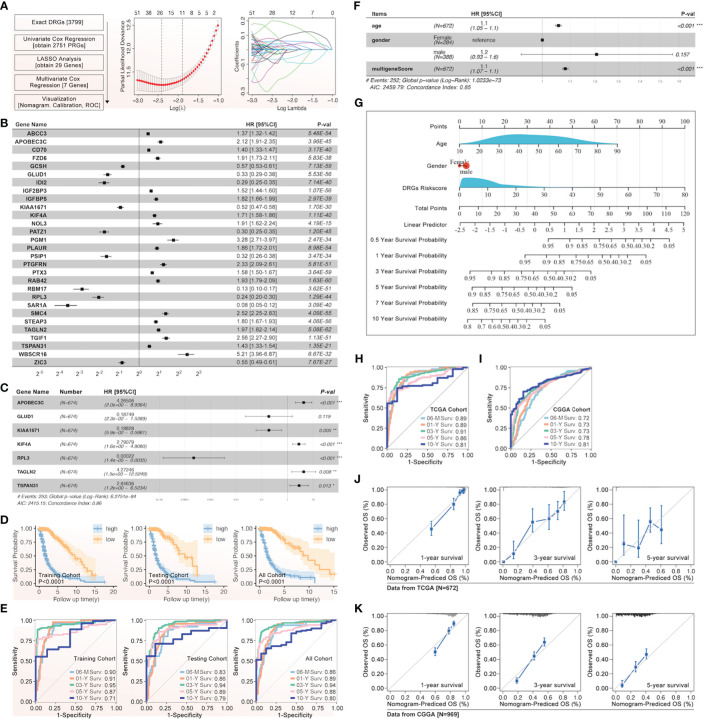
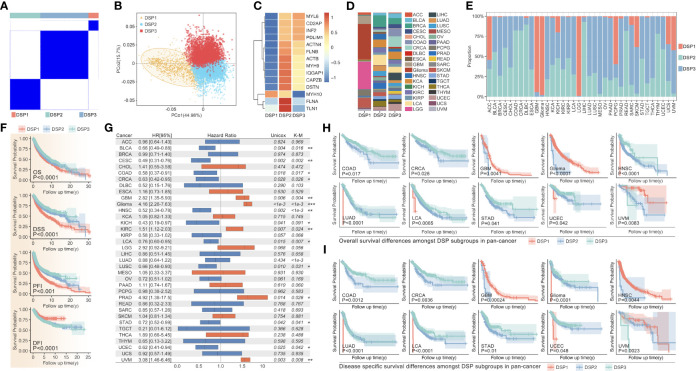
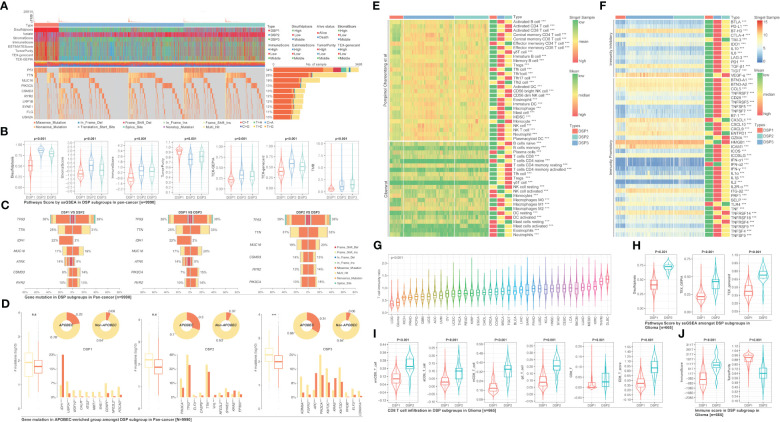
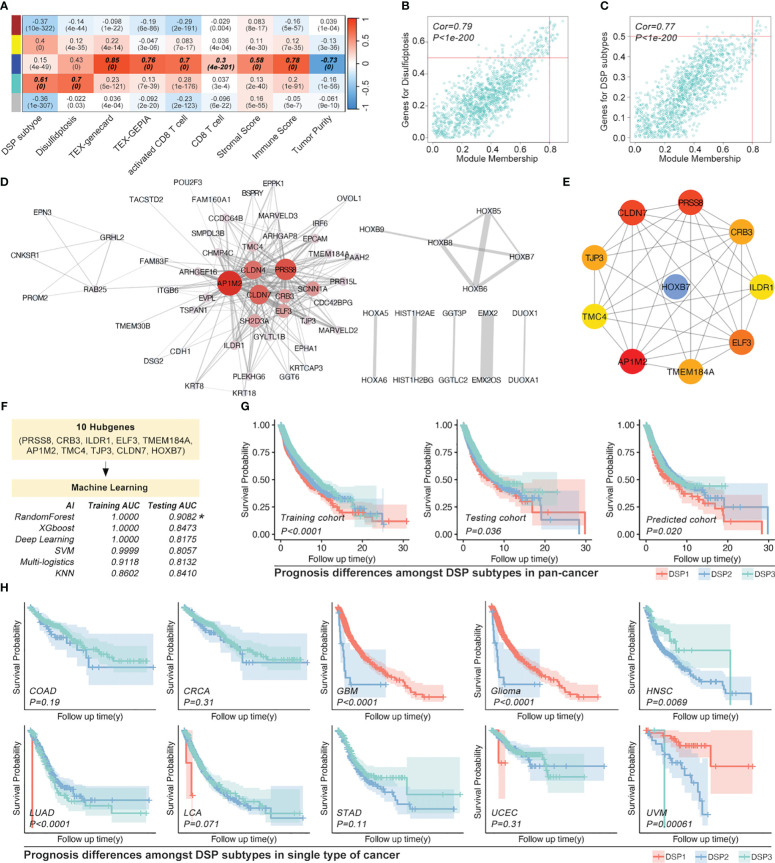
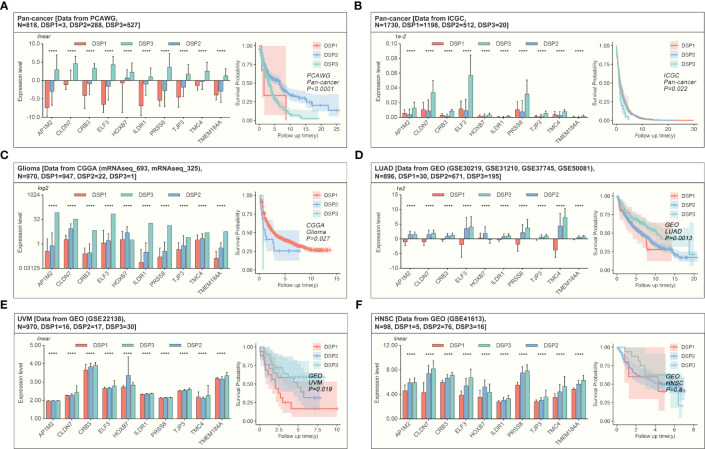
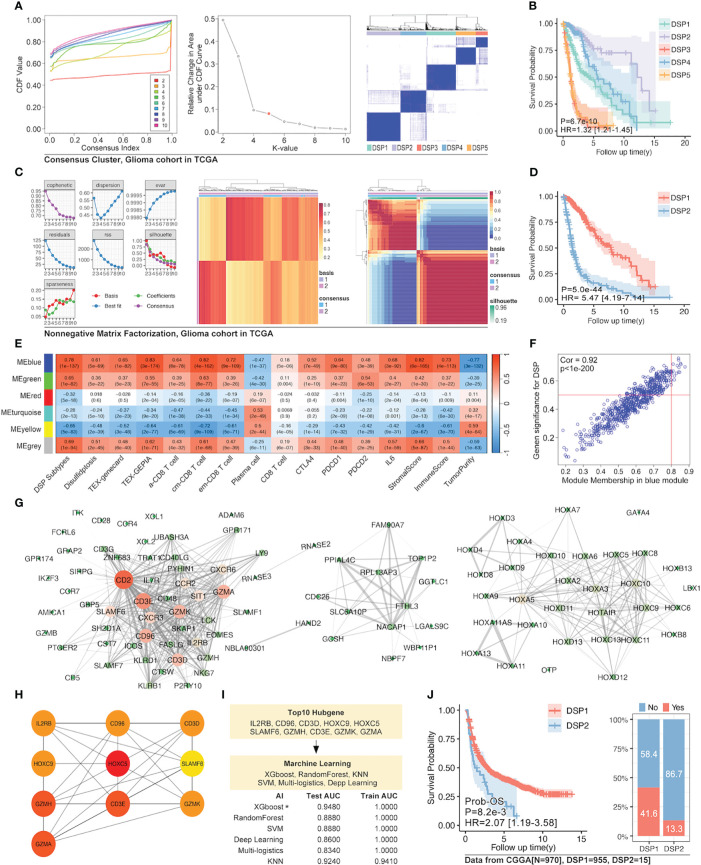
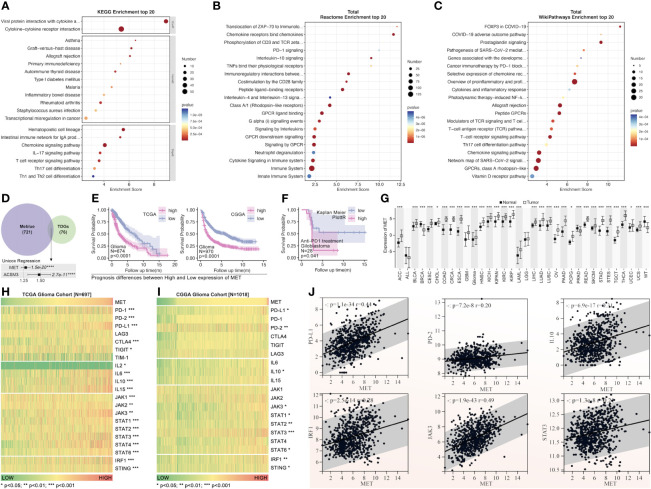
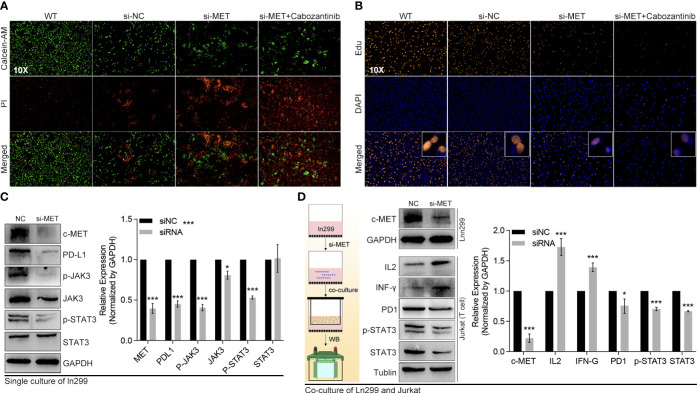
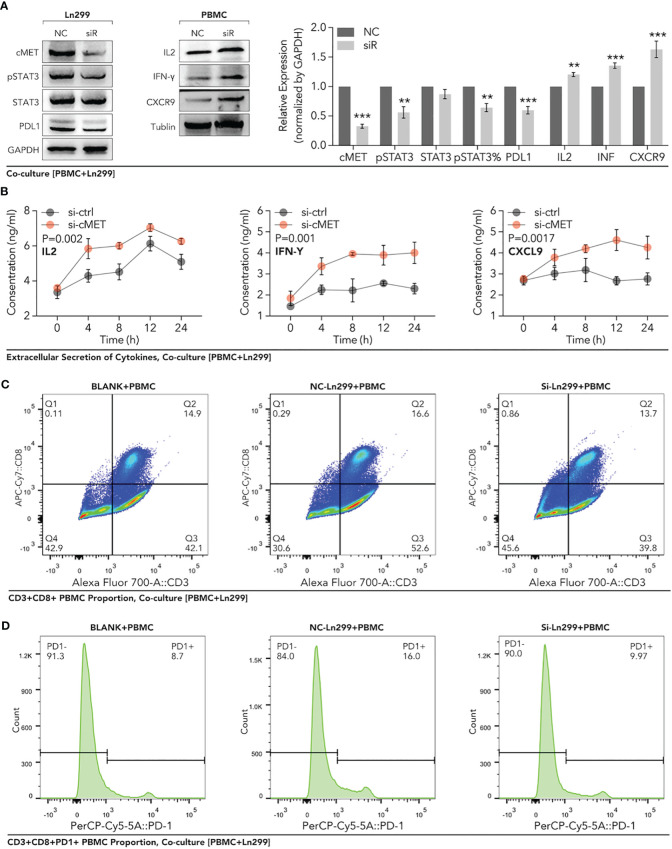
Methods: Based on TCGA, ICGC, CGGA, GSE30219, GSE31210, GSE37745, GSE50081, GSE22138, GSE41613, univariate Cox regression, LASSO regression, and multivariate Cox regression were used to construct the rough gene signature based on disulfidptosis for each type of cancer. SsGSEA and Cibersort, followed by correlation analysis, were harnessed to explore the linkage between disulfidptosis and cancer immunity. Weighted correlation network analysis (WGCNA) and Machine learning were utilized to make a refined prognosis model for pan-cancer. In particular, a customized, enhanced prognosis model was made for glioma. The siRNA transfection, FACS, ELISA, etc., were employed to validate the function of c-MET.
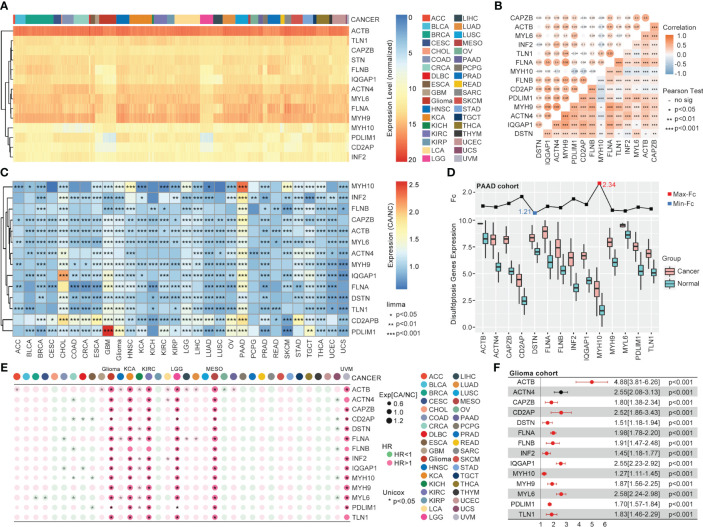
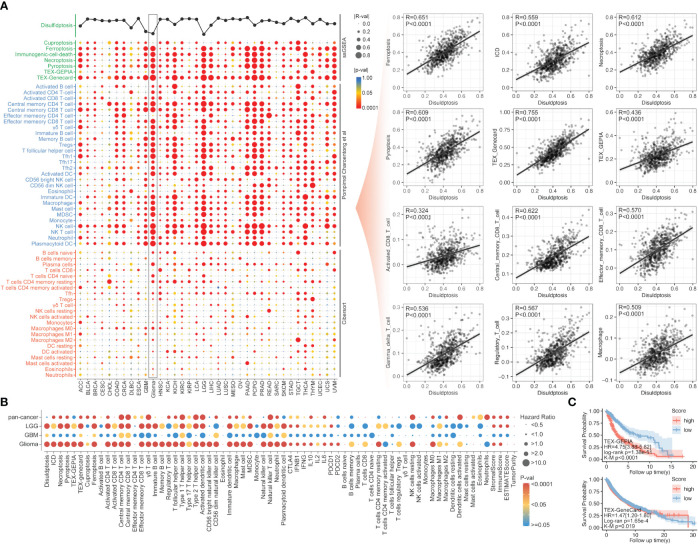
Results: The expression comparison of the disulfidptosis-related genes (DRGs) between tumor and nontumor tissues implied a significant difference in most cancers. The correlation between disulfidptosis and immune cell infiltration, including T cell exhaustion (Tex), was evident, especially in glioma. The 7-gene signature was constructed as the rough model for the glioma prognosis. A pan-cancer suitable DSP clustering was made and validated to predict the prognosis. Furthermore, two DSP groups were defined by machine learning to predict the survival and immune therapy response in glioma, which was validated in CGGA. PD-L1 and other immune pathways were highly enriched in the core blue gene module from WGCNA. Among them, c-MET was validated as a tumor driver gene and JAK3-STAT3-PD-L1/PD1 regulator in glioma and T cells. Specifically, the down-regulation of c-MET decreased the proportion of PD1+ CD8+ T cells.
Conclusion: To summarize, we dissected the roles of DRGs in the prognosis and their relationship with immunity in pan-cancer. A general prognosis model based on machine learning was constructed for pan-cancer and validated by external datasets with a consistent result. In particular, a survival-predicting model was made specifically for patients with glioma to predict its survival and immune response to ICIs. C-MET was screened and validated for its tumor driver gene and immune regulation function (inducing t-cell exhaustion) in glioma.
Keywords: artificial intelligence (AI); disulfidptosis; glioma; prognosis prediction; tumor immunity.
Copyright © 2024 Li, Wang, Wan, Huang, Yin, Sun, Jin and Wang.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous