

A rapid and versatile reverse genetics approach for generating recombinant positive-strand RNA viruses that use IRES-mediated translation
- PMID: 38353536
- PMCID: PMC10949505
- DOI: 10.1128/jvi.01638-23
A rapid and versatile reverse genetics approach for generating recombinant positive-strand RNA viruses that use IRES-mediated translation
Abstract
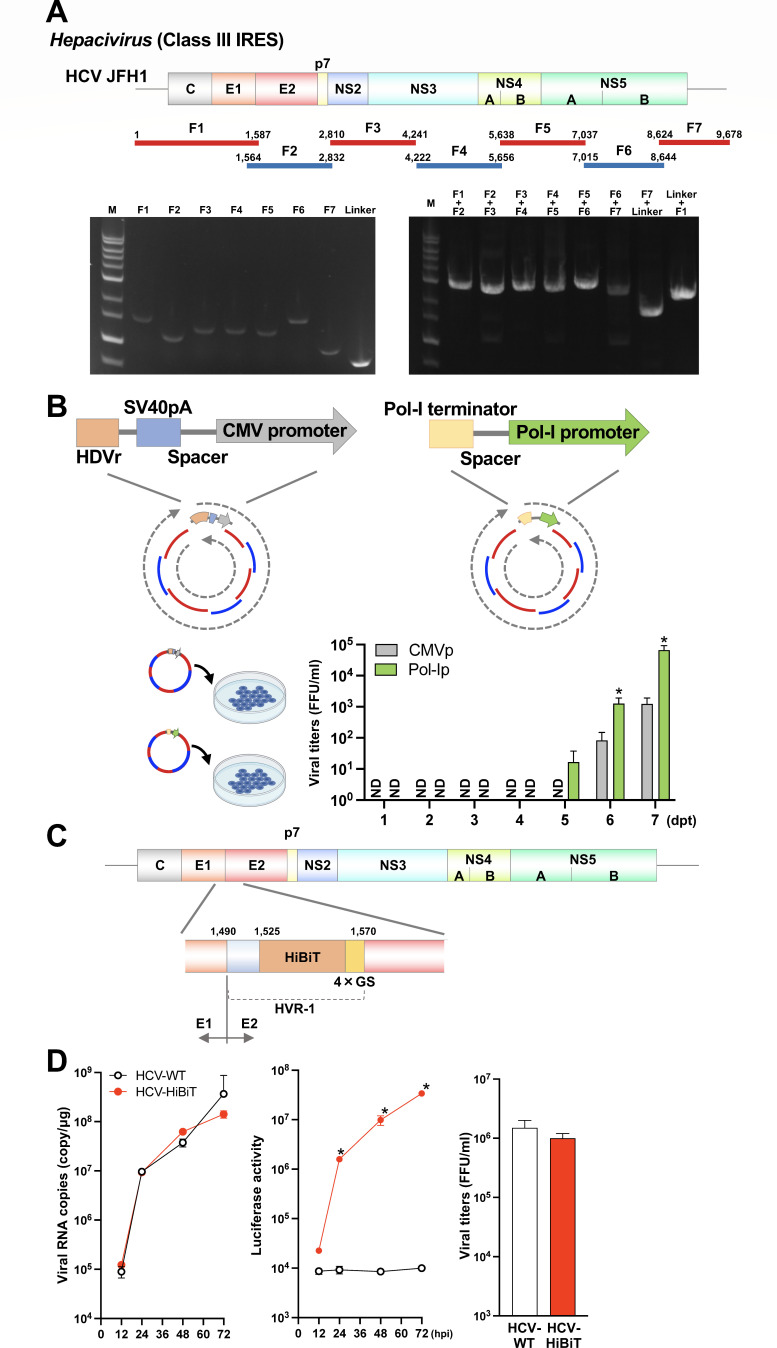
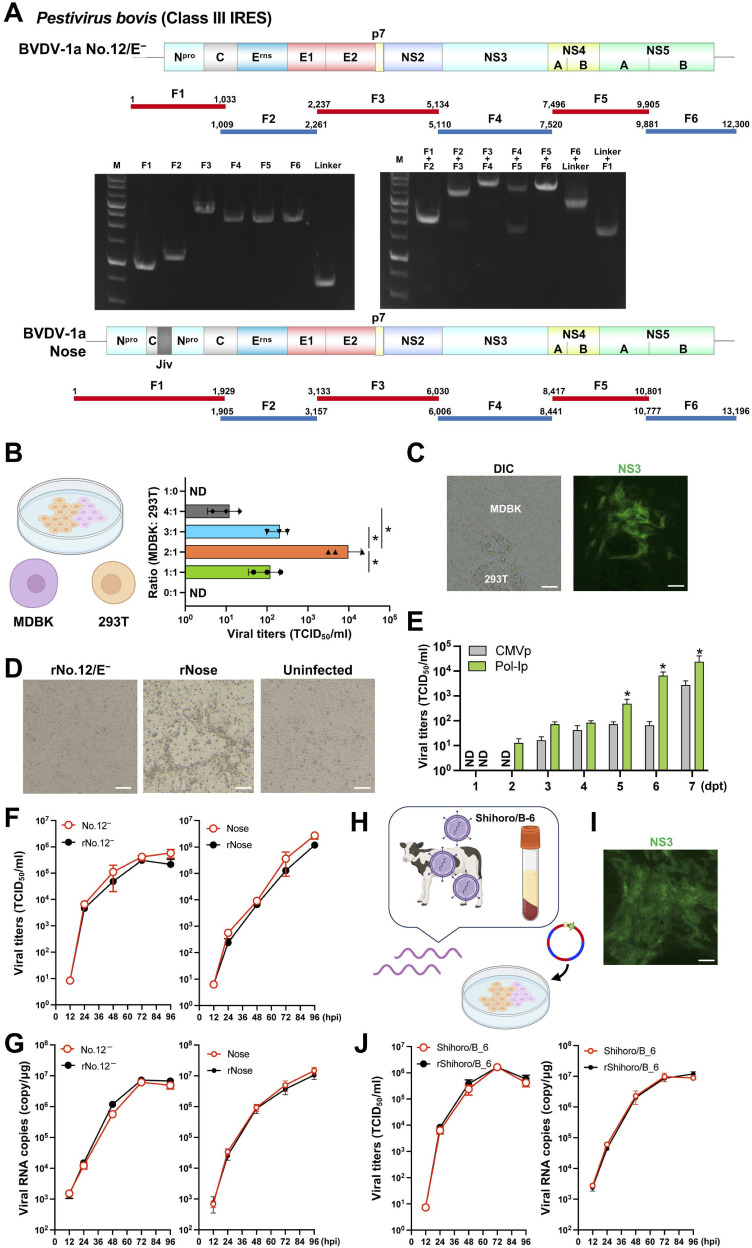
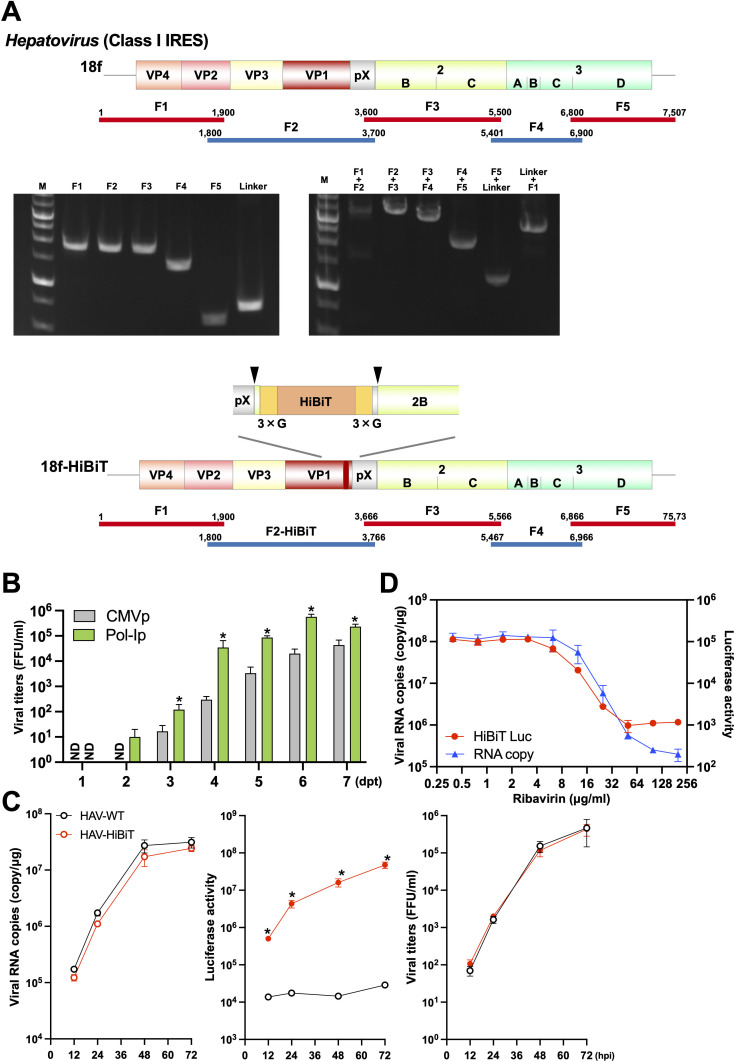
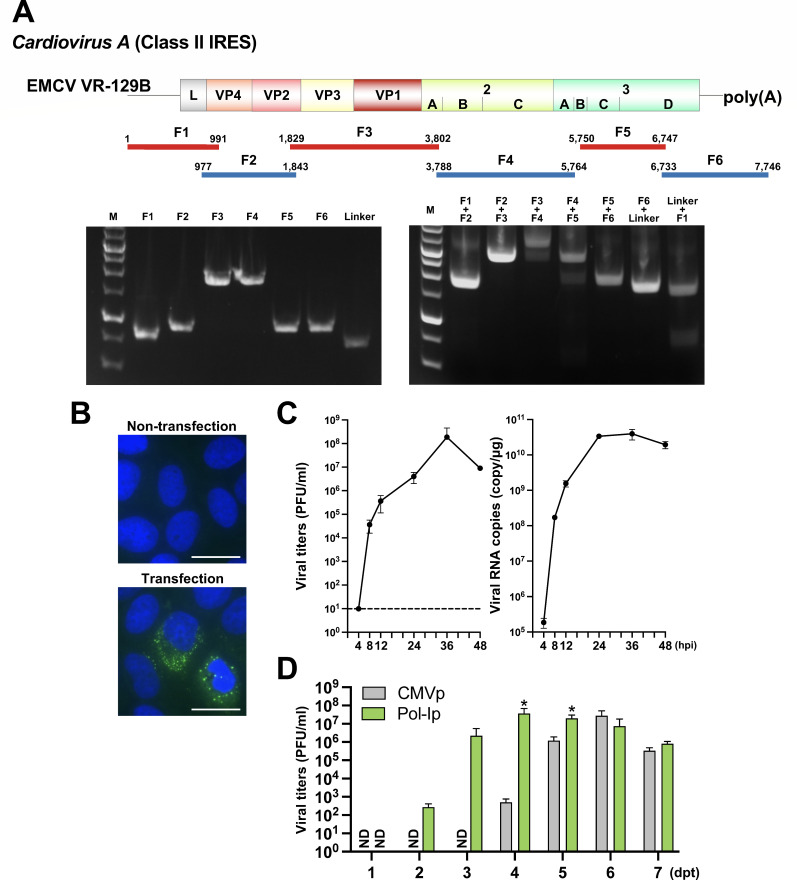
Reverse genetics systems have played a central role in developing recombinant viruses for a wide spectrum of virus research. The circular polymerase extension reaction (CPER) method has been applied to studying positive-strand RNA viruses, allowing researchers to bypass molecular cloning of viral cDNA clones and thus leading to the rapid generation of recombinant viruses. However, thus far, the CPER protocol has only been established using cap-dependent RNA viruses. Here, we demonstrate that a modified version of the CPER method can be successfully applied to positive-strand RNA viruses that use cap-independent, internal ribosomal entry site (IRES)-mediated translation. As a proof-of-concept, we employed mammalian viruses with different types (classes I, II, and III) of IRES to optimize the CPER method. Using the hepatitis C virus (HCV, class III), we found that inclusion in the CPER assembly of an RNA polymerase I promoter and terminator, instead of those from polymerase II, allowed greater viral production. This approach was also successful in generating recombinant bovine viral diarrhea virus (class III) following transfection of MDBK/293T co-cultures to overcome low transfection efficiency. In addition, we successfully generated the recombinant viruses from clinical specimens. Our modified CPER could be used for producing hepatitis A virus (HAV, type I) as well as de novo generation of encephalomyocarditis virus (type II). Finally, we generated recombinant HCV and HAV reporter viruses that exhibited replication comparable to that of the wild-type parental viruses. The recombinant HAV reporter virus helped evaluate antivirals. Taking the findings together, this study offers methodological advances in virology.
Importance: The lack of versatility of reverse genetics systems remains a bottleneck in viral research. Especially when (re-)emerging viruses reach pandemic levels, rapid characterization and establishment of effective countermeasures using recombinant viruses are beneficial in disease control. Indeed, numerous studies have attempted to establish and improve the methods. The circular polymerase extension reaction (CPER) method has overcome major obstacles in generating recombinant viruses. However, this method has not yet been examined for positive-strand RNA viruses that use cap-independent, internal ribosome entry site-mediated translation. Here, we engineered a suitable gene cassette to expand the CPER method for all positive-strand RNA viruses. Furthermore, we overcame the difficulty of generating recombinant viruses because of low transfection efficiency. Using this modified method, we also successfully generated reporter viruses and recombinant viruses from a field sample without virus isolation. Taking these findings together, our adapted methodology is an innovative technology that could help advance virologic research.
Keywords: IRES; RNA virus; cap-independence; reporter virus; reverse genetics.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Yeh MT, Bujaki E, Dolan PT, Smith M, Wahid R, Konz J, Weiner AJ, Bandyopadhyay AS, Van Damme P, De Coster I, Revets H, Macadam A, Andino R. 2020. Engineering the live-attenuated polio vaccine to prevent reversion to virulence. Cell Host Microbe 27:736–751. doi: 10.1016/j.chom.2020.04.003 - DOI - PMC - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
