

Genetic determinants of micronucleus formation in vivo
- PMID: 38355793
- PMCID: PMC10917660
- DOI: 10.1038/s41586-023-07009-0
Genetic determinants of micronucleus formation in vivo
Abstract
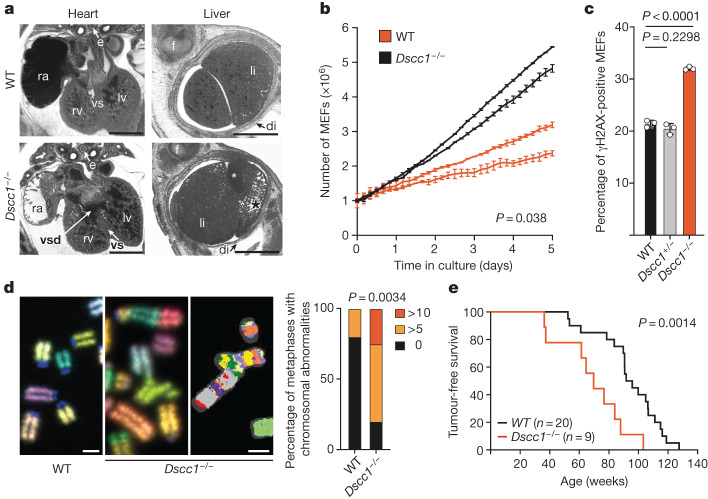
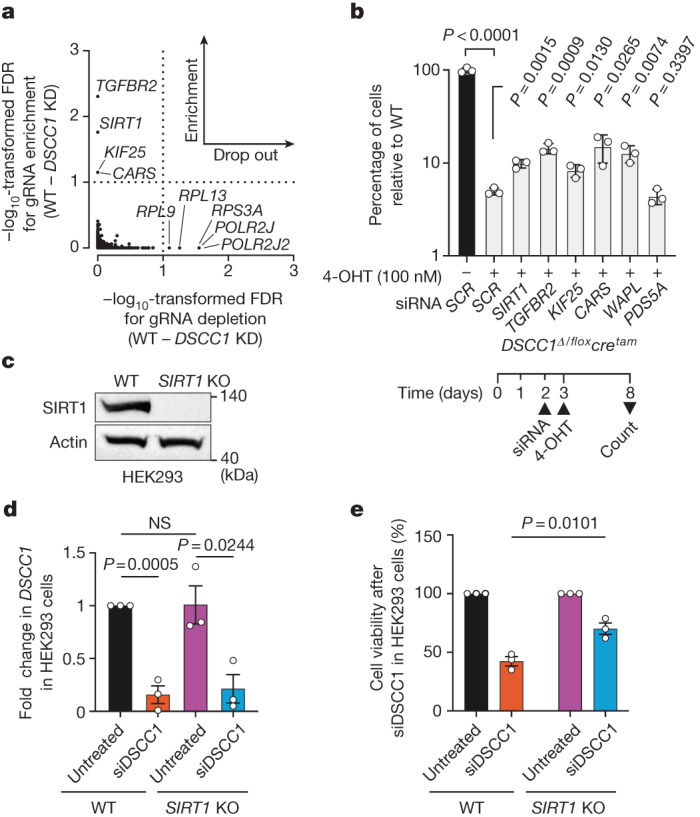
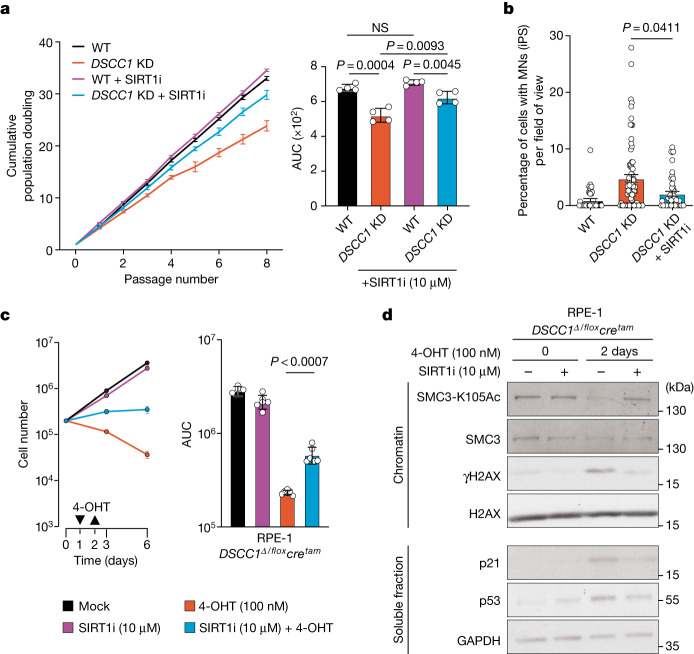
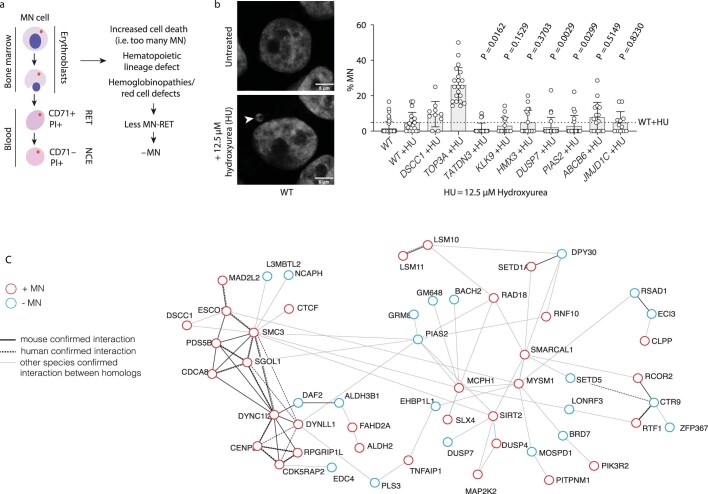
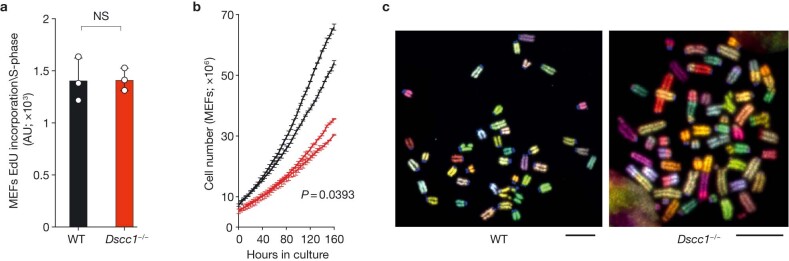
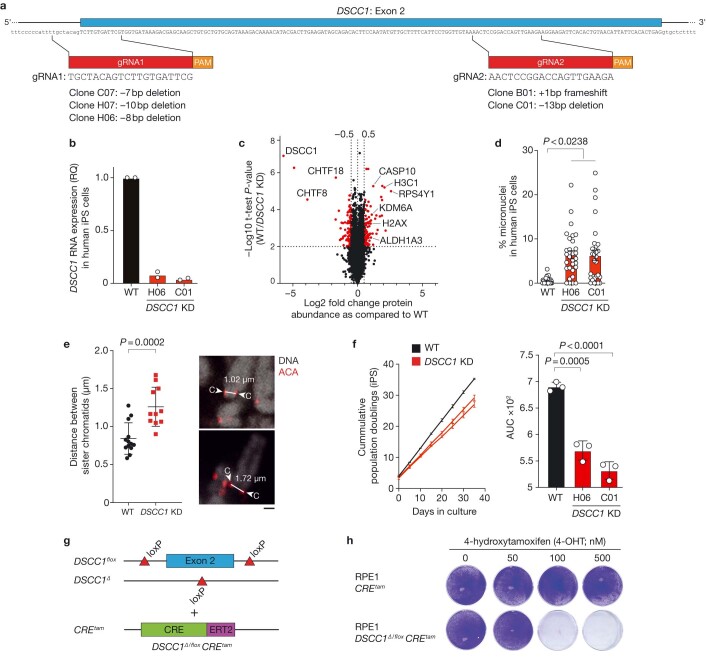
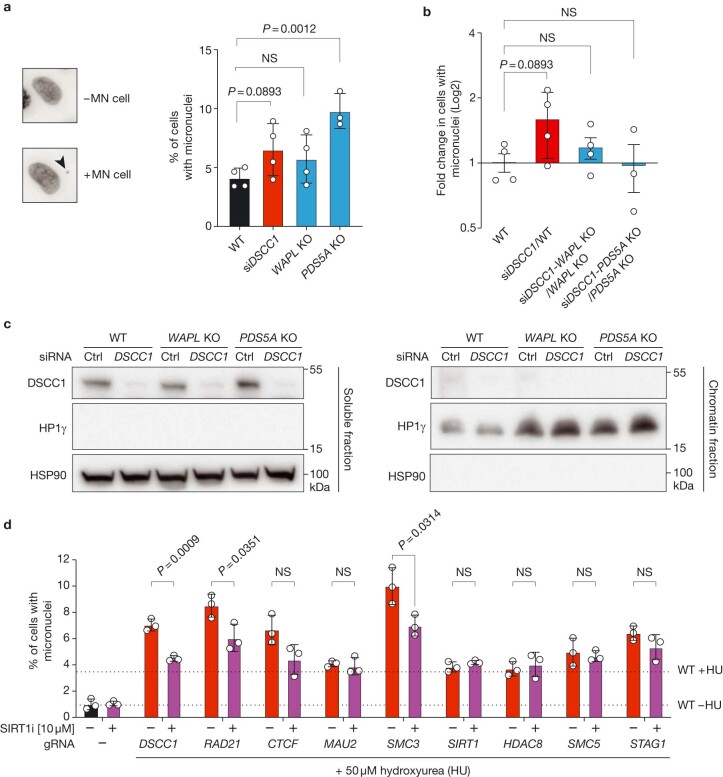
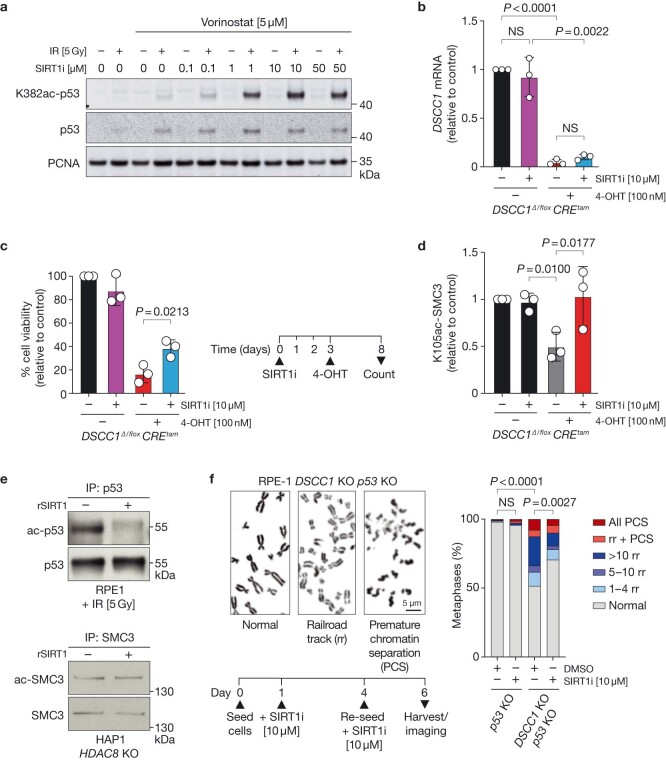
Genomic instability arising from defective responses to DNA damage1 or mitotic chromosomal imbalances2 can lead to the sequestration of DNA in aberrant extranuclear structures called micronuclei (MN). Although MN are a hallmark of ageing and diseases associated with genomic instability, the catalogue of genetic players that regulate the generation of MN remains to be determined. Here we analyse 997 mouse mutant lines, revealing 145 genes whose loss significantly increases (n = 71) or decreases (n = 74) MN formation, including many genes whose orthologues are linked to human disease. We found that mice null for Dscc1, which showed the most significant increase in MN, also displayed a range of phenotypes characteristic of patients with cohesinopathy disorders. After validating the DSCC1-associated MN instability phenotype in human cells, we used genome-wide CRISPR-Cas9 screening to define synthetic lethal and synthetic rescue interactors. We found that the loss of SIRT1 can rescue phenotypes associated with DSCC1 loss in a manner paralleling restoration of protein acetylation of SMC3. Our study reveals factors involved in maintaining genomic stability and shows how this information can be used to identify mechanisms that are relevant to human disease biology1.
© 2024. The Author(s).
Conflict of interest statement
A patent on the repurposing of SIRT1 inhibitors has been filed by the University of Cambridge. The data presented in this patent are included in the main paper and Supplementary Information. D.J.A. is a consultant for Microbiotica and Ono Therapeutics and receives research funding from Astra Zeneca and OpenTargets. M.R. is employed by Artios Pharma and is also a shareholder. J.R.B.P. is an employee of Insmed Innovation UK and holds stock/stock options in Insmed, and also receives research funding from GSK. The other authors declare no competing interests.
Figures

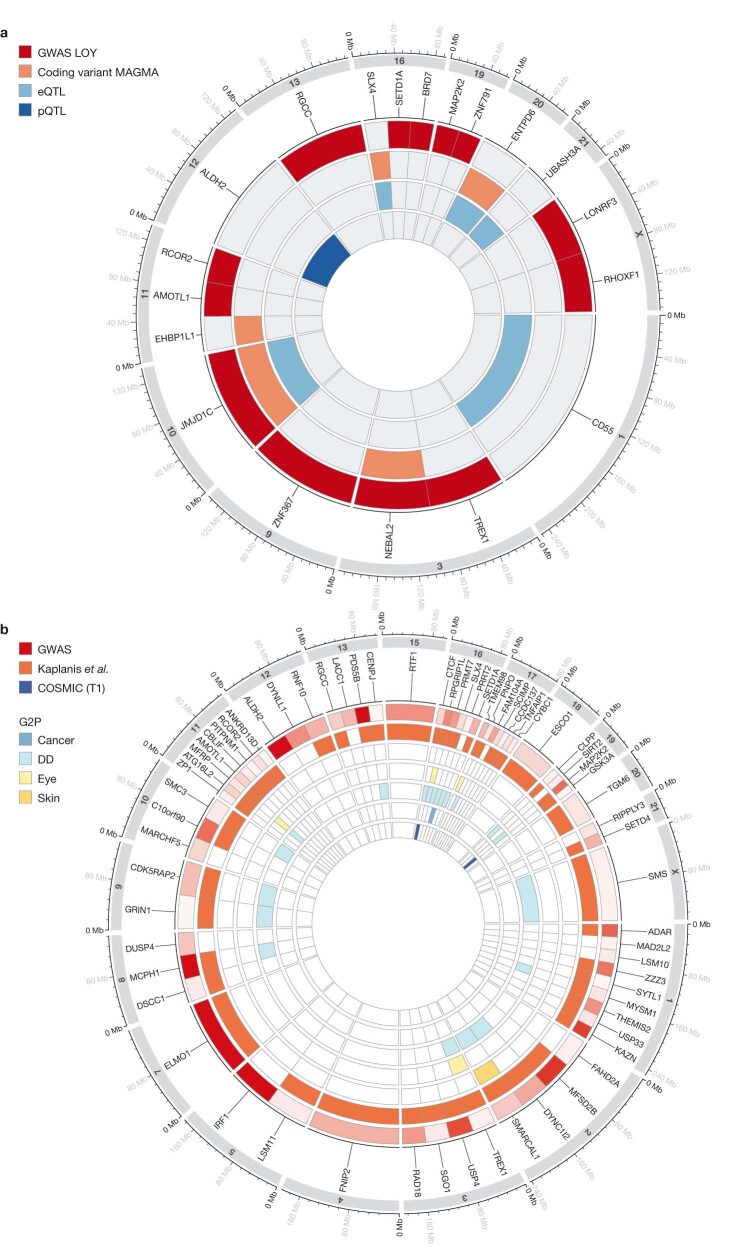
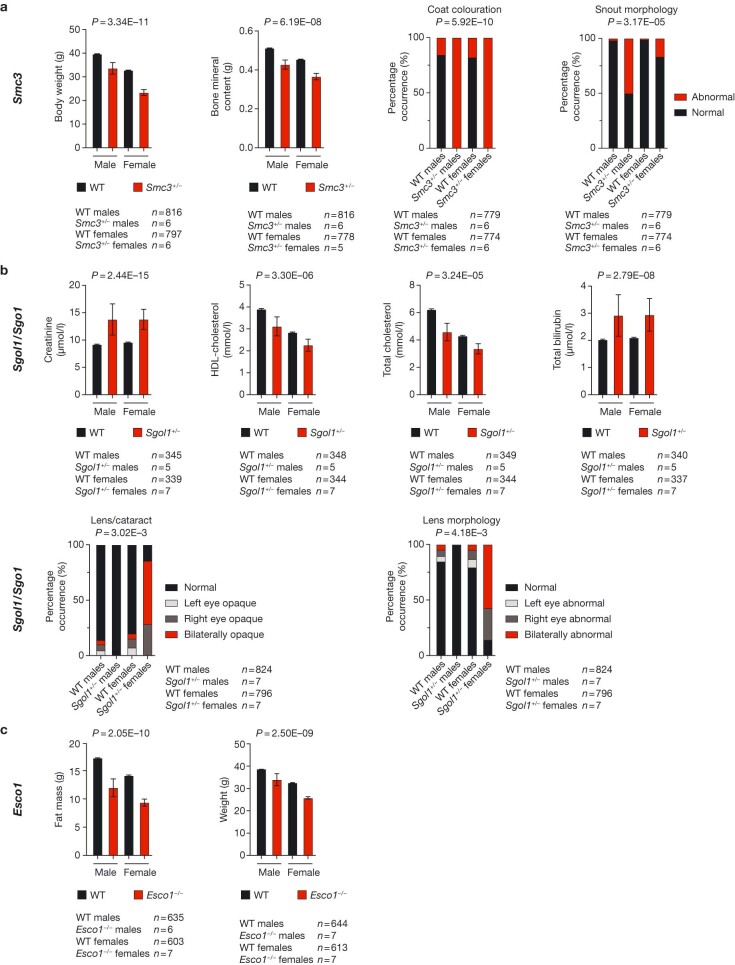
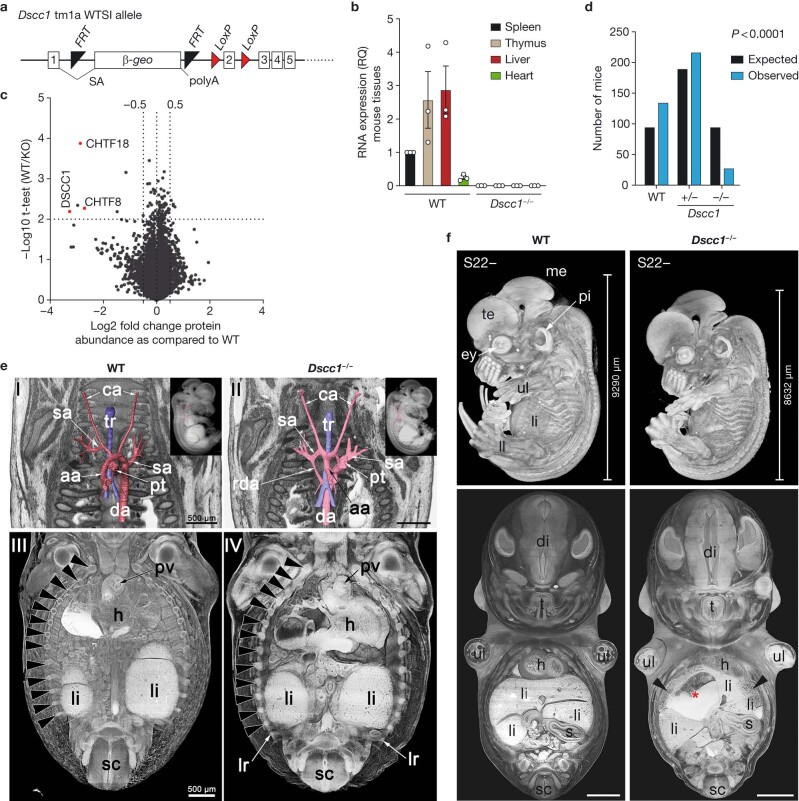
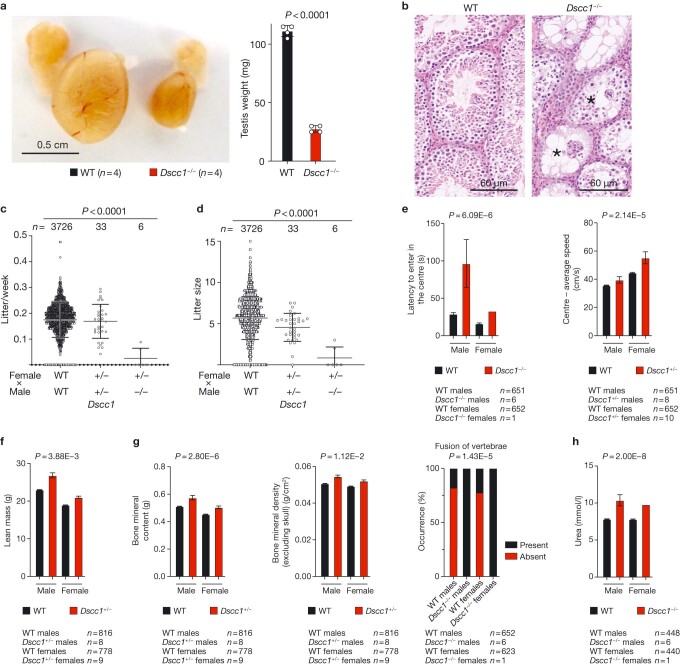
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
