

A single-cell time-lapse of mouse prenatal development from gastrula to birth
- PMID: 38355799
- PMCID: PMC10901739
- DOI: 10.1038/s41586-024-07069-w
A single-cell time-lapse of mouse prenatal development from gastrula to birth
Abstract
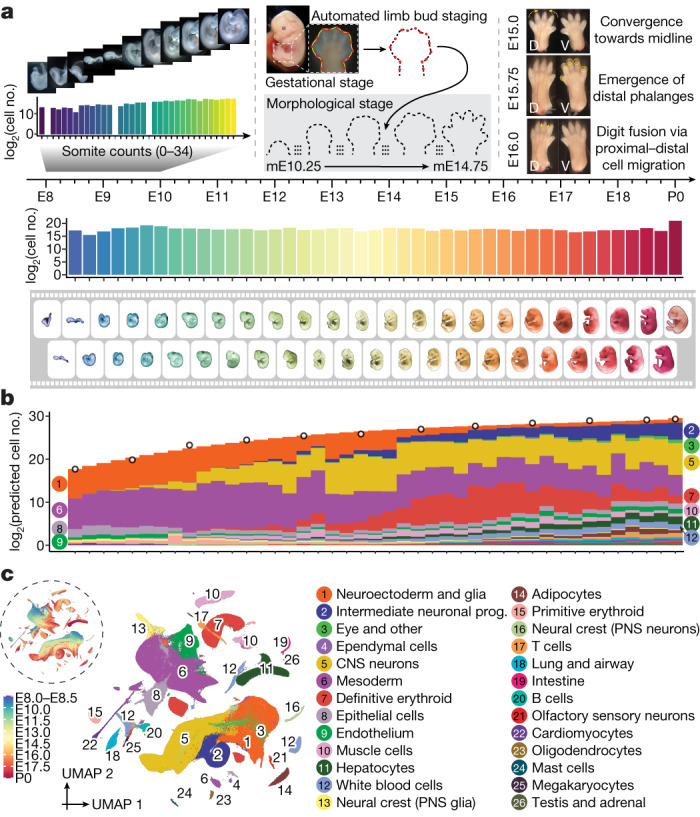
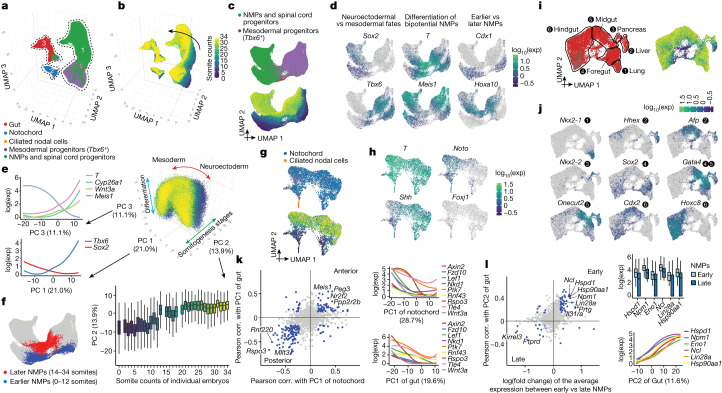
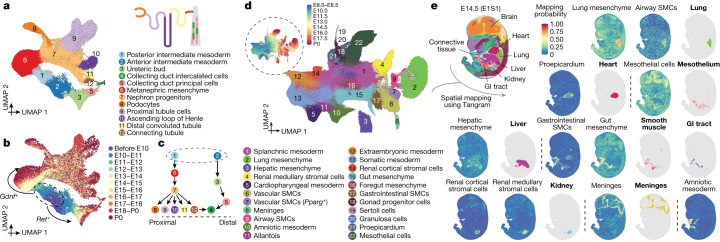
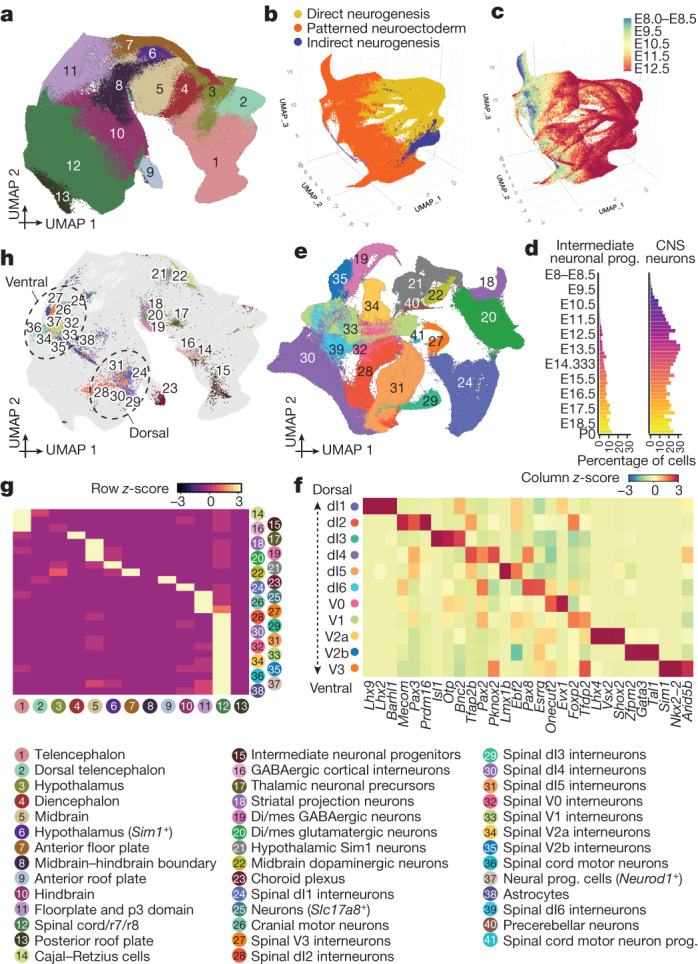
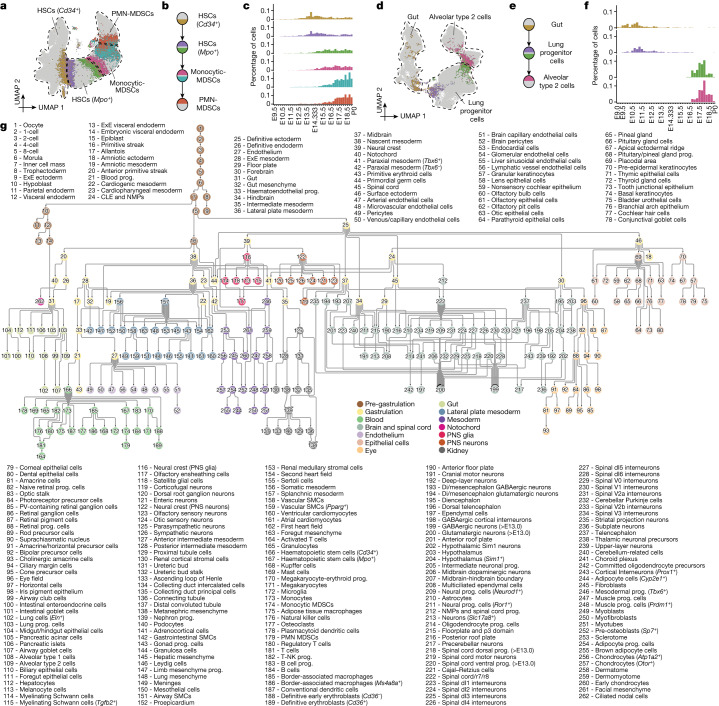
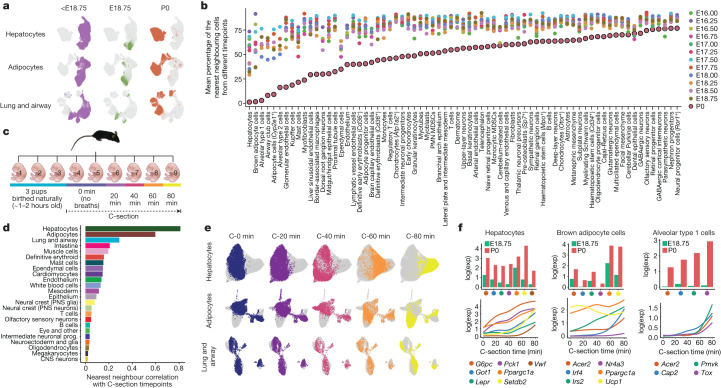
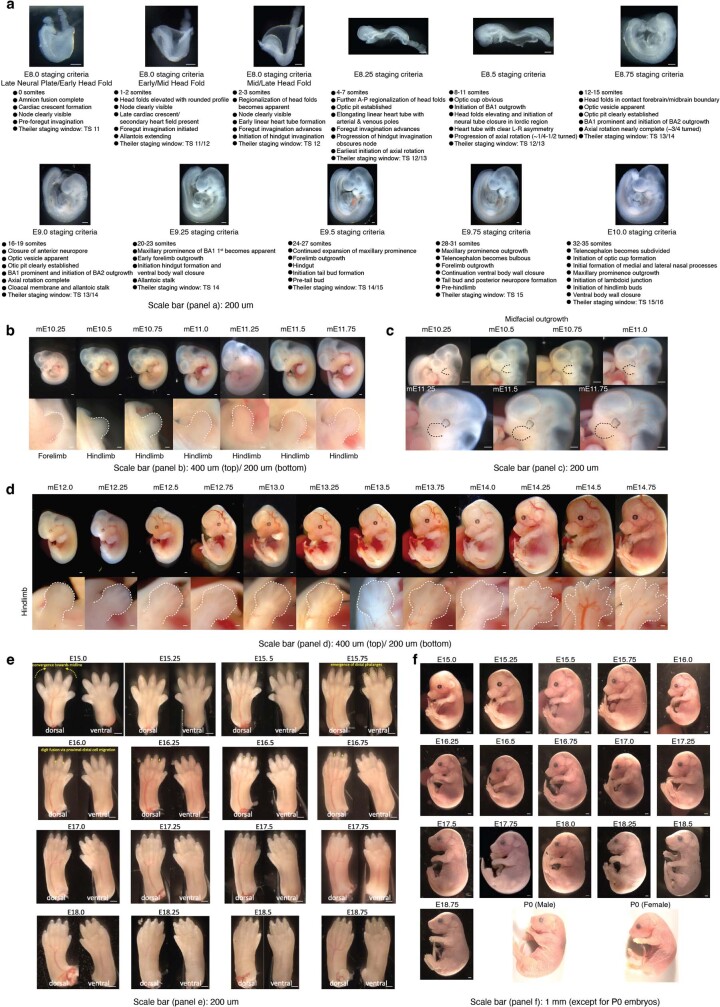
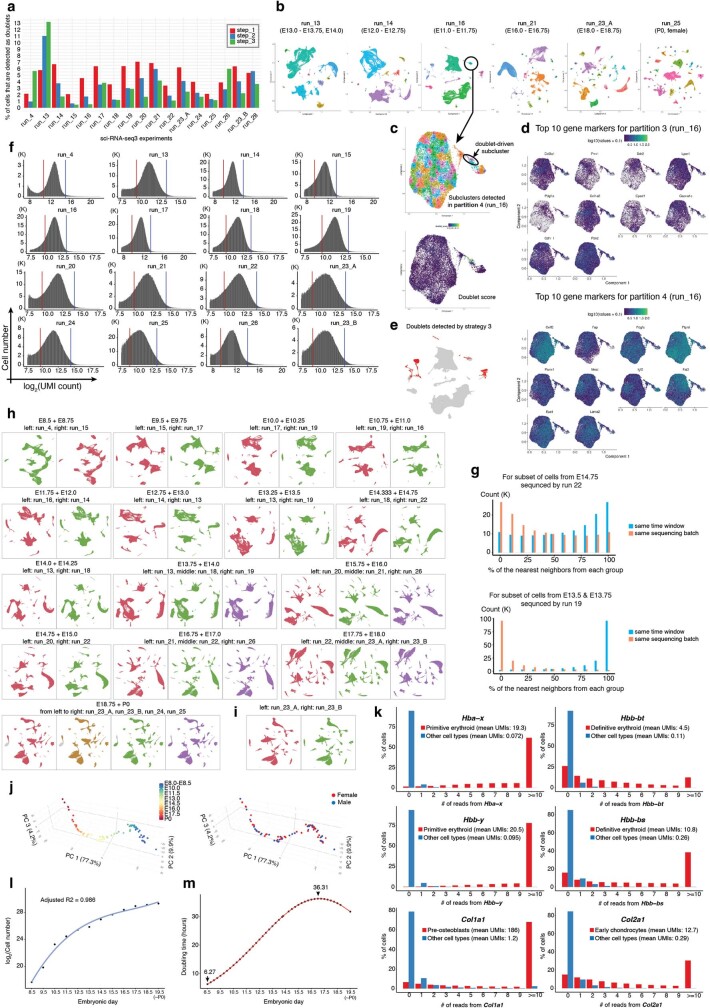
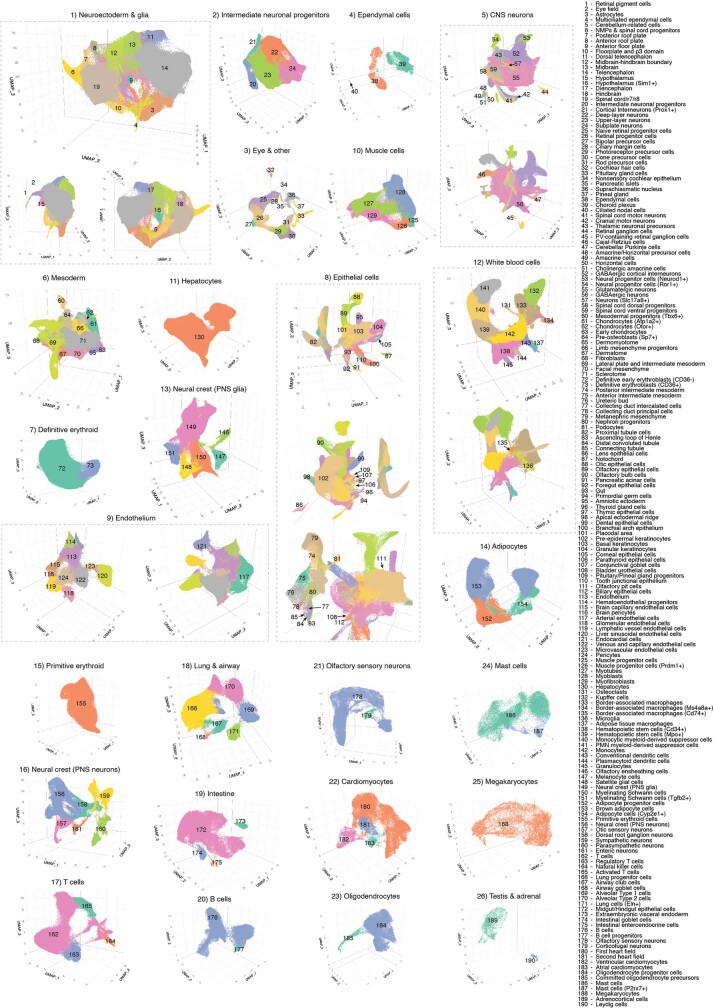
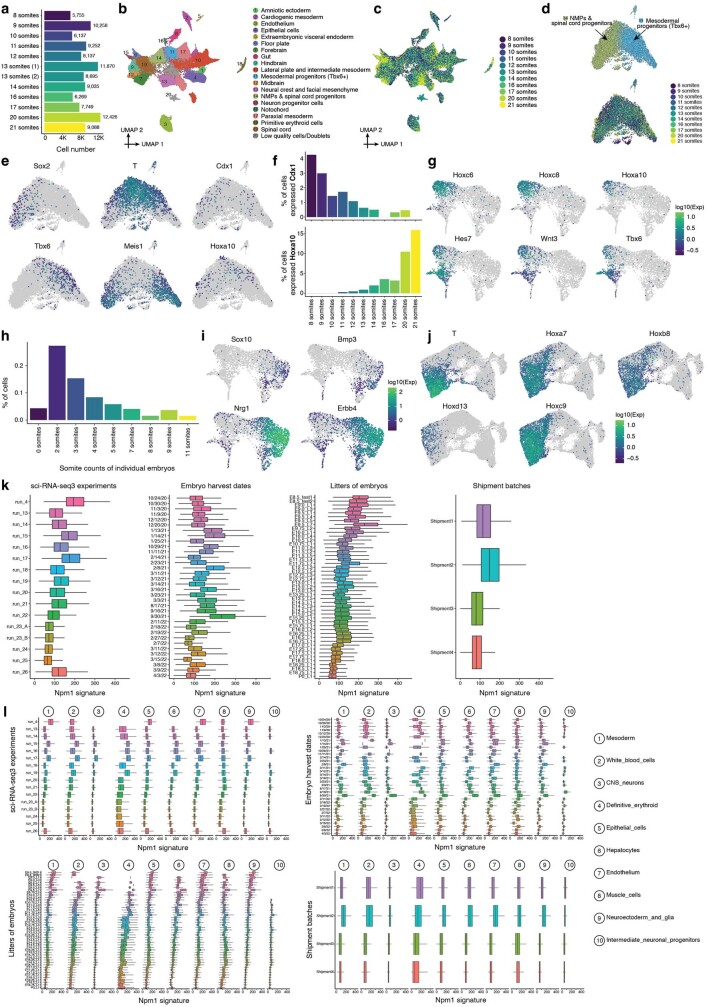
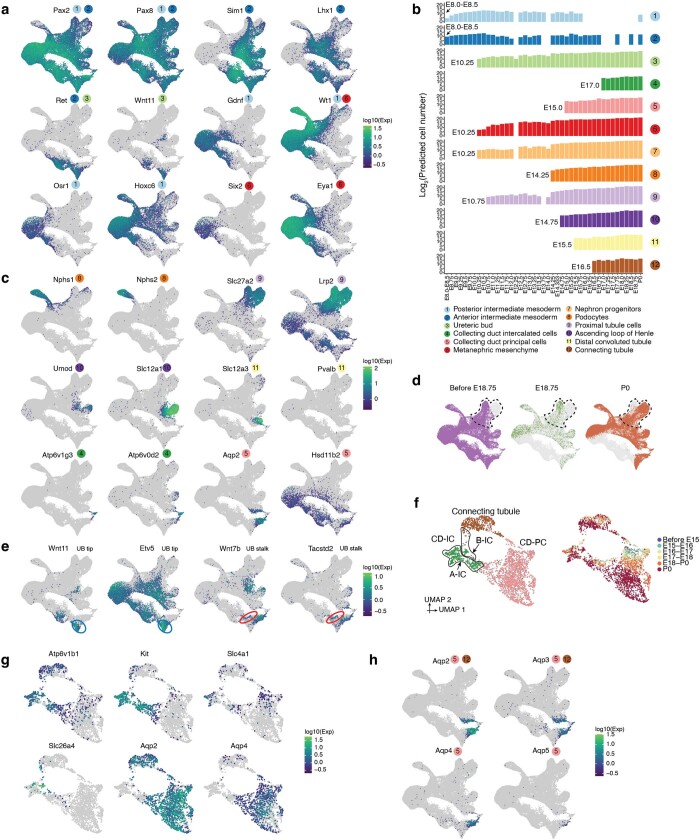
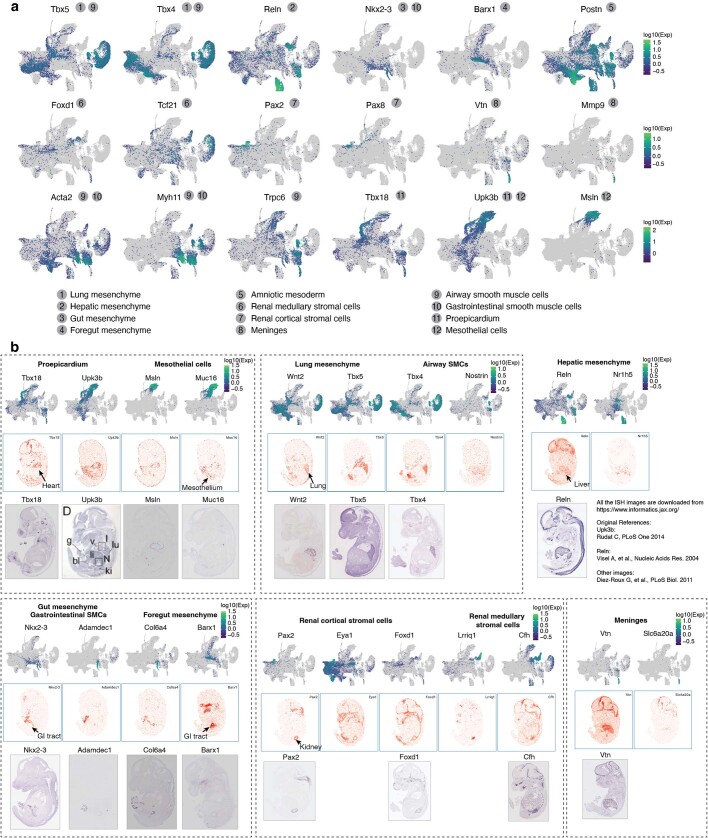
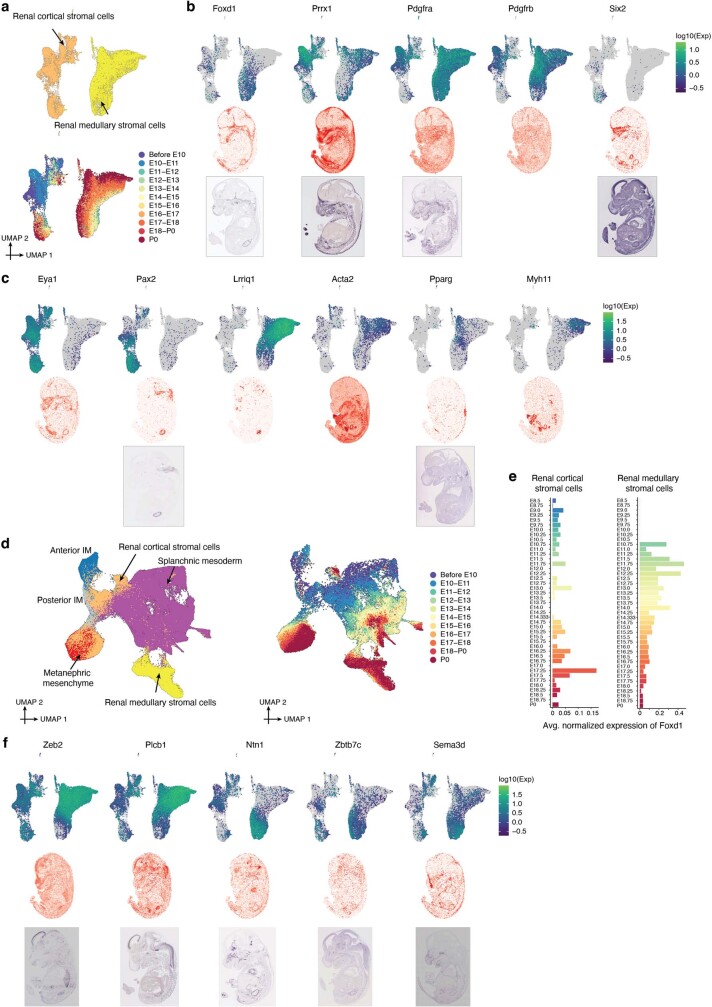
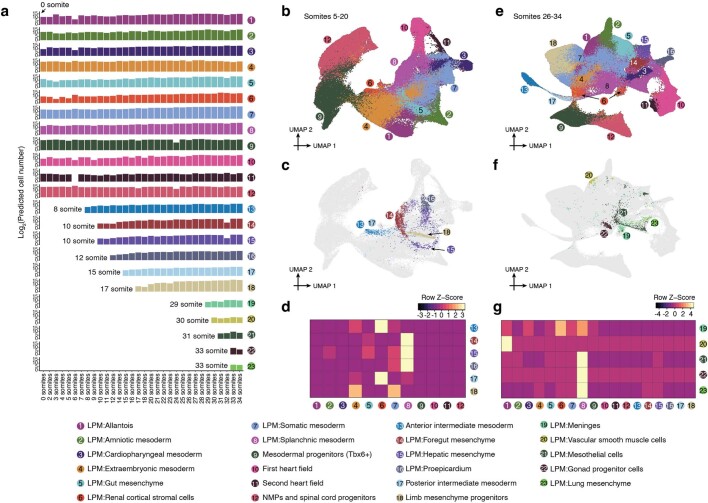
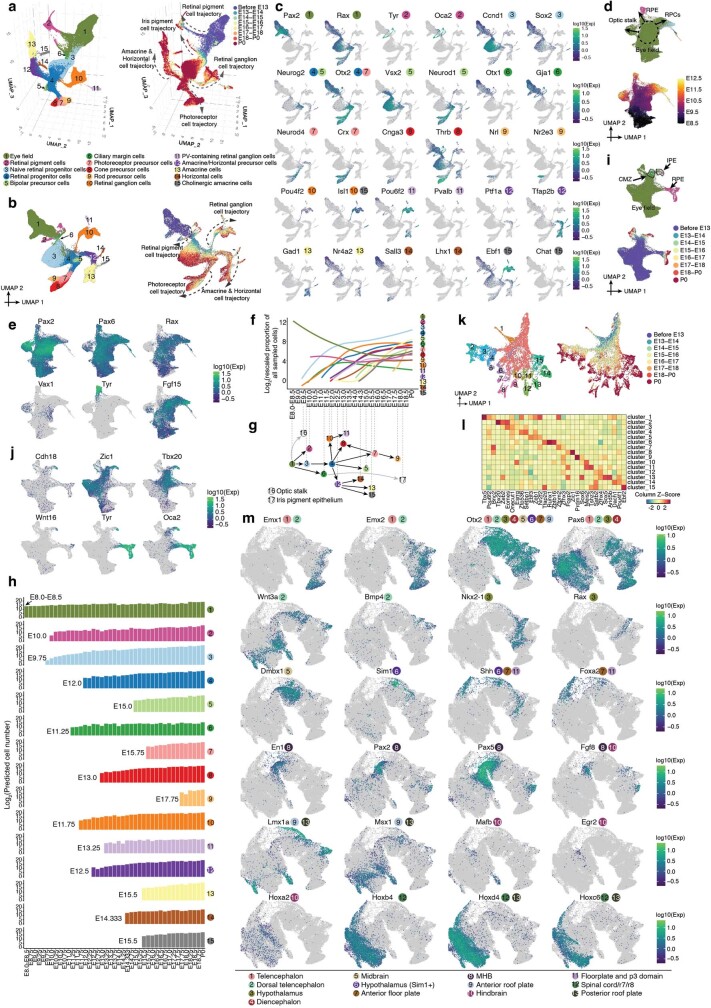
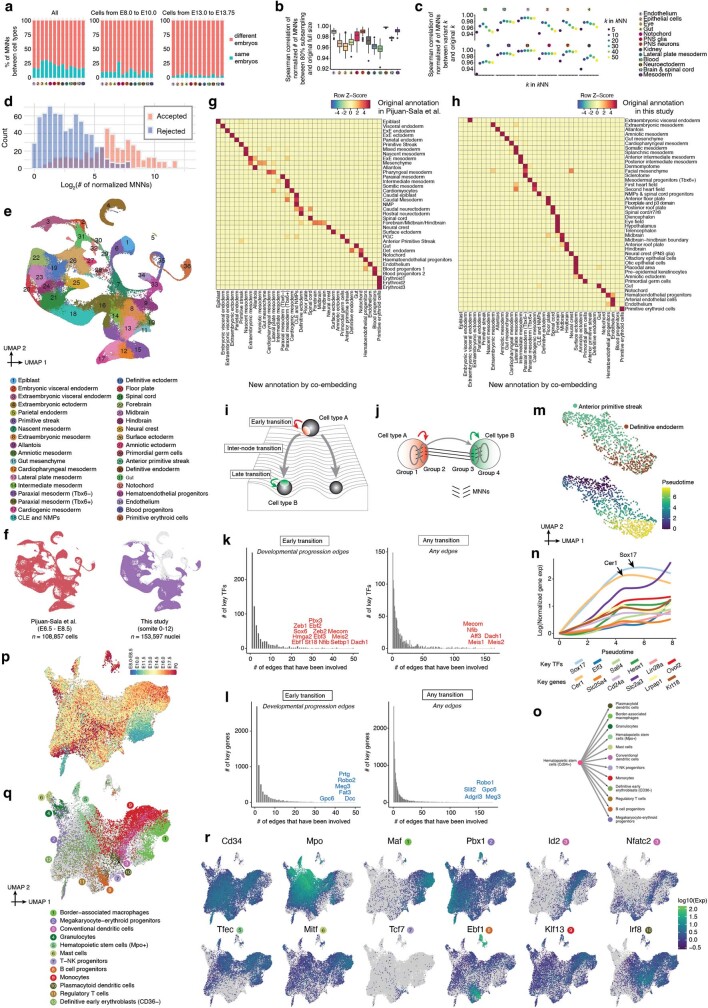
The house mouse (Mus musculus) is an exceptional model system, combining genetic tractability with close evolutionary affinity to humans1,2. Mouse gestation lasts only 3 weeks, during which the genome orchestrates the astonishing transformation of a single-cell zygote into a free-living pup composed of more than 500 million cells. Here, to establish a global framework for exploring mammalian development, we applied optimized single-cell combinatorial indexing3 to profile the transcriptional states of 12.4 million nuclei from 83 embryos, precisely staged at 2- to 6-hour intervals spanning late gastrulation (embryonic day 8) to birth (postnatal day 0). From these data, we annotate hundreds of cell types and explore the ontogenesis of the posterior embryo during somitogenesis and of kidney, mesenchyme, retina and early neurons. We leverage the temporal resolution and sampling depth of these whole-embryo snapshots, together with published data4-8 from earlier timepoints, to construct a rooted tree of cell-type relationships that spans the entirety of prenatal development, from zygote to birth. Throughout this tree, we systematically nominate genes encoding transcription factors and other proteins as candidate drivers of the in vivo differentiation of hundreds of cell types. Remarkably, the most marked temporal shifts in cell states are observed within one hour of birth and presumably underlie the massive physiological adaptations that must accompany the successful transition of a mammalian fetus to life outside the womb.
© 2024. The Author(s).
Conflict of interest statement
J.S. and C.T. are co-founders and scientific advisors to Scale Biosciences. J.S. is also a scientific advisory board member, consultant and/or co-founder of Cajal Neuroscience, Guardant Health, Maze Therapeutics, Camp4 Therapeutics, Phase Genomics, Adaptive Biotechnologies, Sixth Street Capital, Pacific Biosciences, and Prime Medicine. The other authors declare no competing interests.
Figures

Update of
-
A single-cell transcriptional timelapse of mouse embryonic development, from gastrula to pup.bioRxiv [Preprint]. 2023 Apr 5:2023.04.05.535726. doi: 10.1101/2023.04.05.535726. bioRxiv. 2023. Update in: Nature. 2024 Feb;626(8001):1084-1093. doi: 10.1038/s41586-024-07069-w. PMID: 37066300 Free PMC article. Updated. Preprint.
References
-
- Silver, L. M. Mouse Genetics: Concepts and Applications (Oxford Univ. Press, 1995).
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
