

Efanesoctocog alfa: the renaissance of Factor VIII replacement therapy
- PMID: 38356459
- PMCID: PMC11290510
- DOI: 10.3324/haematol.2023.284498
Efanesoctocog alfa: the renaissance of Factor VIII replacement therapy
Abstract
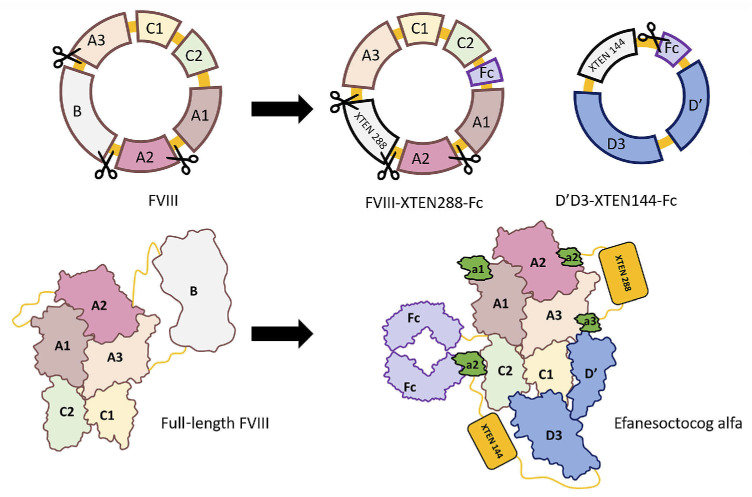
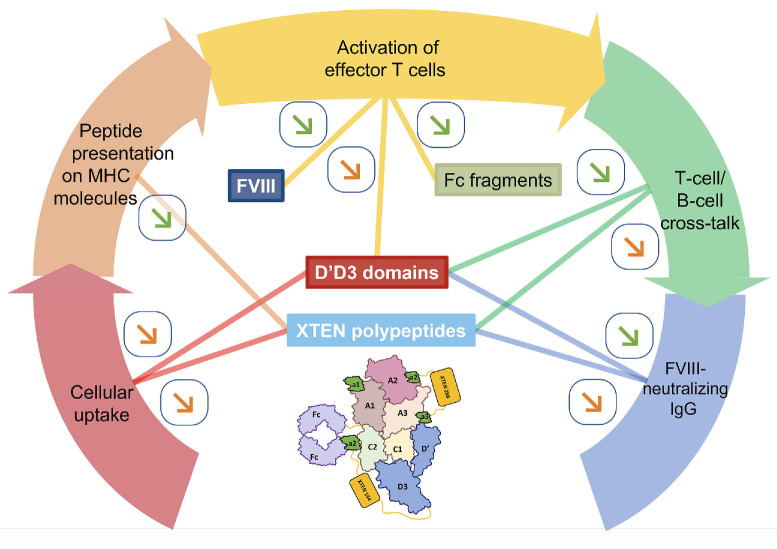
Efanesoctocog alfa (Altuviiio,TM Sanofi-SOBI) is a B domain-deleted single-chain Factor VIII (FVIII) connected to D'D3 domain of von Willebrand Factor (vWF). Its ingenious design allows efanesoctocog alfa to operate independently of endogenous vWF and results in an outstanding 3-4 times longer half-life compared to standard and extended half-life (EHL) FVIII products. The prolonged half-life ensures sustained high levels of factor activity, maintaining normal to near-normal ranges for the majority of the week, facilitating the convenience of once-weekly administration. Efanesoctocog alfa received regulatory approval in 2023 for application in both adults and children with inherited hemophilia A in the United States and Japan. Its sanctioned use encompasses both prophylaxis and 'on demand' treatment for bleeding episodes. The European Medicines Agency (EMA) is currently undertaking a comprehensive review of Altuviiio. TM This comprehensive review focuses on the immunological profile of efanesoctocog alfa, a highly sophisticated new class of EHL FVIII molecule. The integration of the vWF D'D3 domain, XTEN polypeptides, and potential regulatory T-cell epitopes within various segments of efanesoctocog alfa collectively serves as a mitigating factor against the development of a neutralizing T-cell-mediated immune response. We hypothesize that such distinctive attribute may significantly reduce the risk of neutralizing antibodies, particularly in previously untreated patients. The discussion extends beyond regulatory approval to encompass the preclinical and clinical development of efanesoctocog alfa, including considerations for laboratory monitoring. The review also highlights areas that warrant further investigation to deepen our understanding of this groundbreaking therapeutic agent.
Figures
References
-
- Mannucci PM, Franchini M. Haematology clinic: haemophilia A. Hematology. 2014;19(3):181-182. - PubMed
-
- Lafeber FP, Miossec P, Valentino LA. Physiopathology of haemophilic arthropathy. Haemophilia. 2008;14(Suppl 4):3-9. - PubMed
-
- Oldenburg J. Optimal treatment strategies for hemophilia: achievements and limitations of current prophylactic regimens. Blood. 2015;125(13):2038-2044. - PubMed
-
- Mannucci PM. Hemophilia treatment innovation: 50 years of progress and more to come. J Thromb Haemost. 2023;21(3):403-412. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
