

Enterovirus A71 does not meet the uncoating receptor SCARB2 at the cell surface
- PMID: 38359079
- PMCID: PMC10901359
- DOI: 10.1371/journal.ppat.1012022
Enterovirus A71 does not meet the uncoating receptor SCARB2 at the cell surface
Abstract
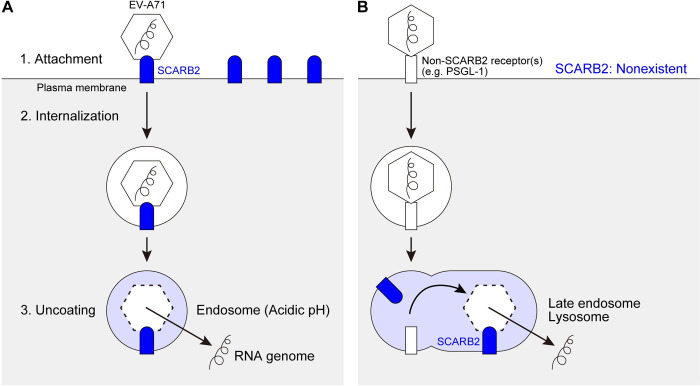
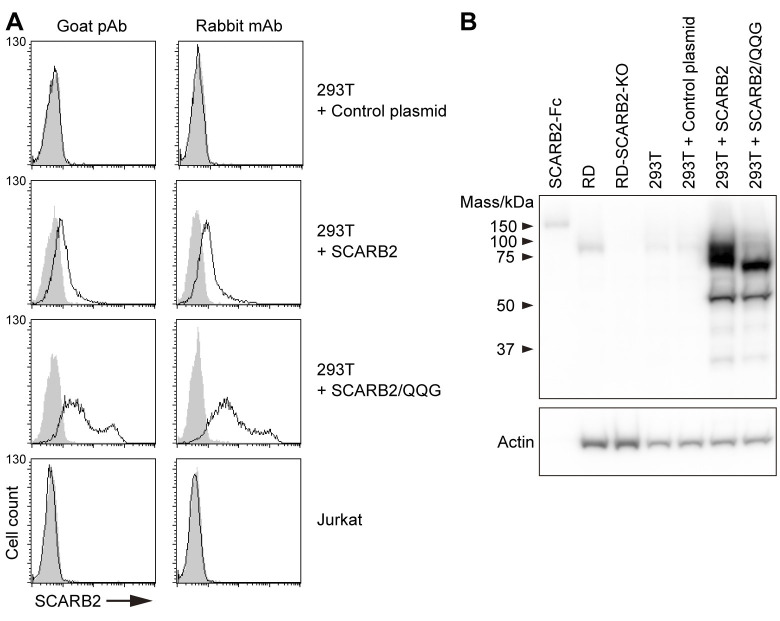
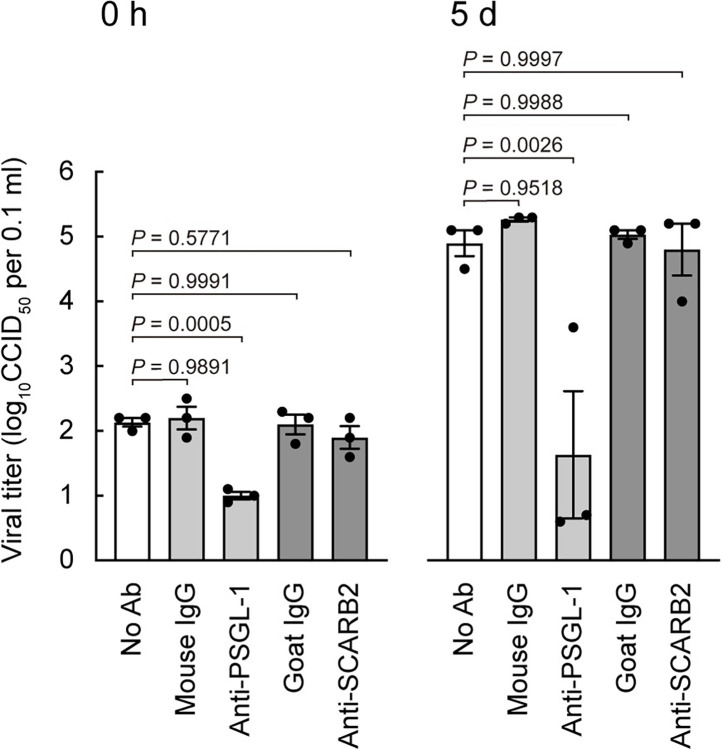
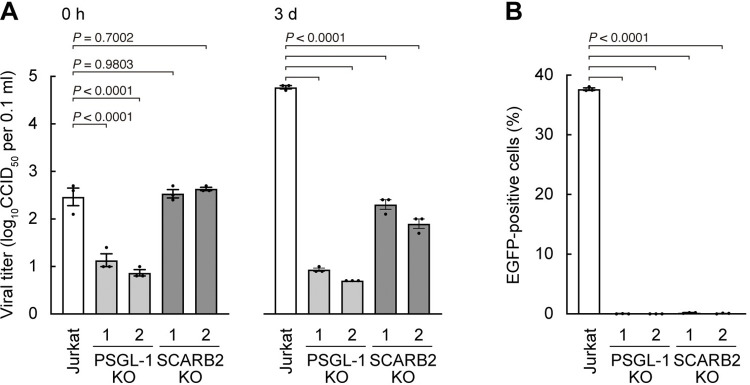
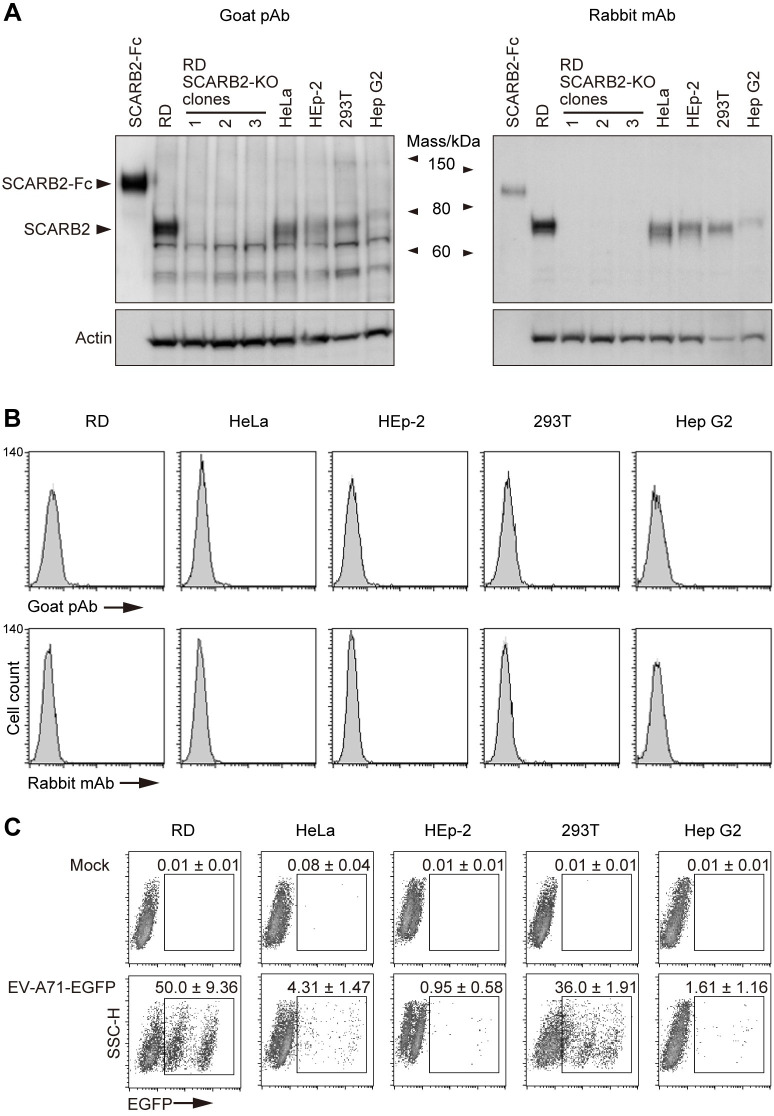
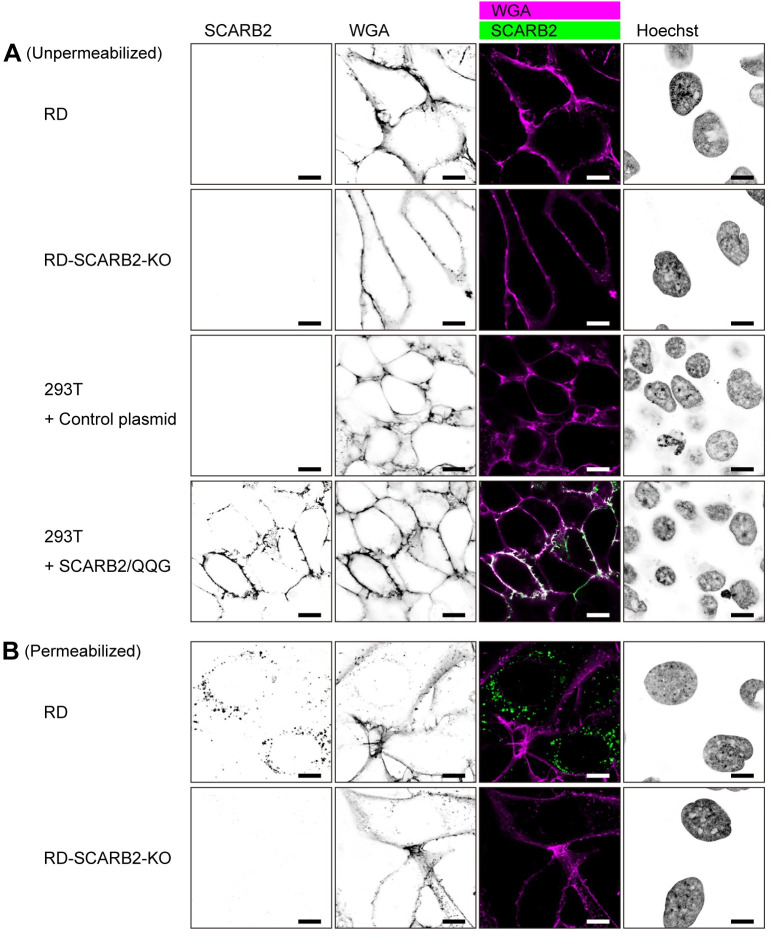
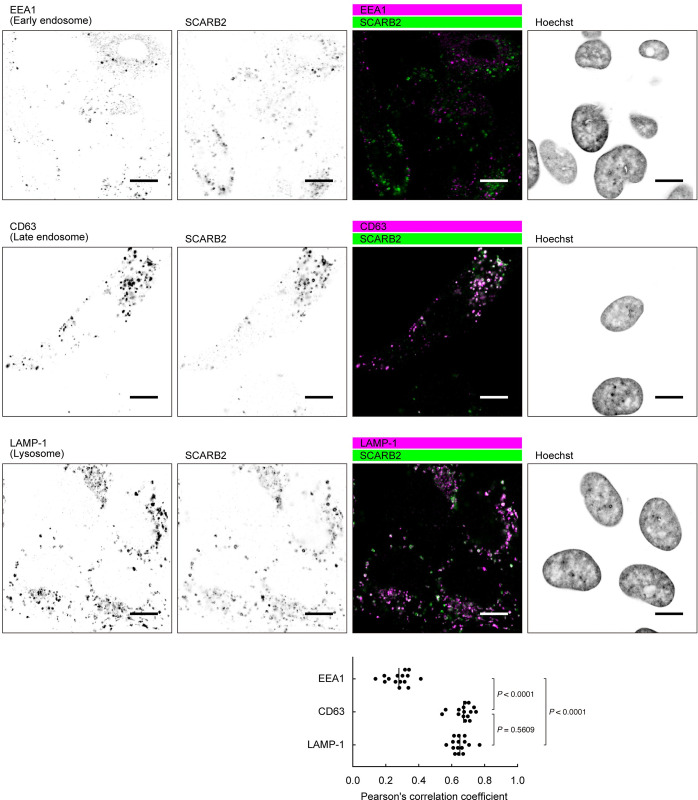
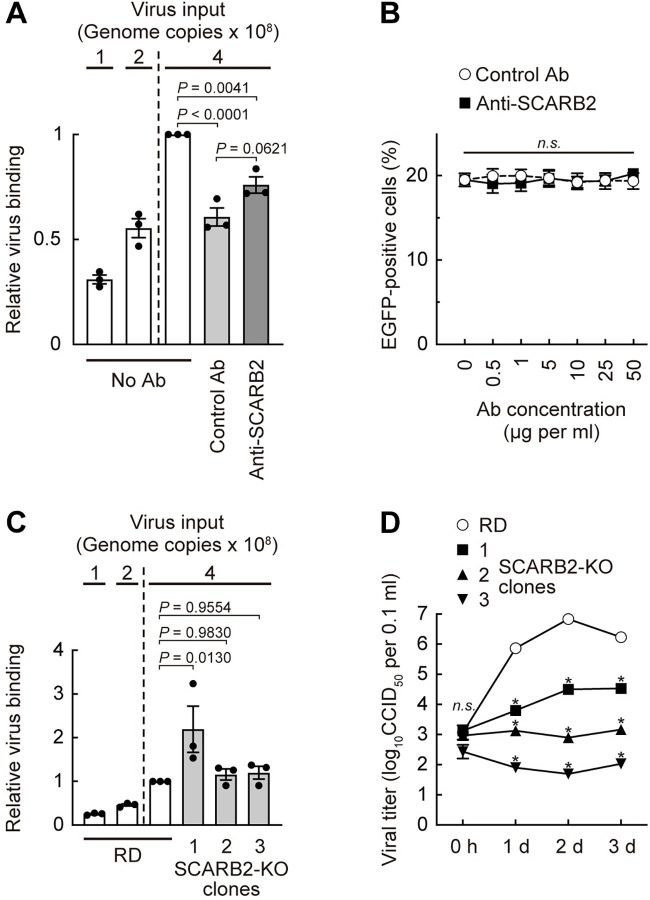
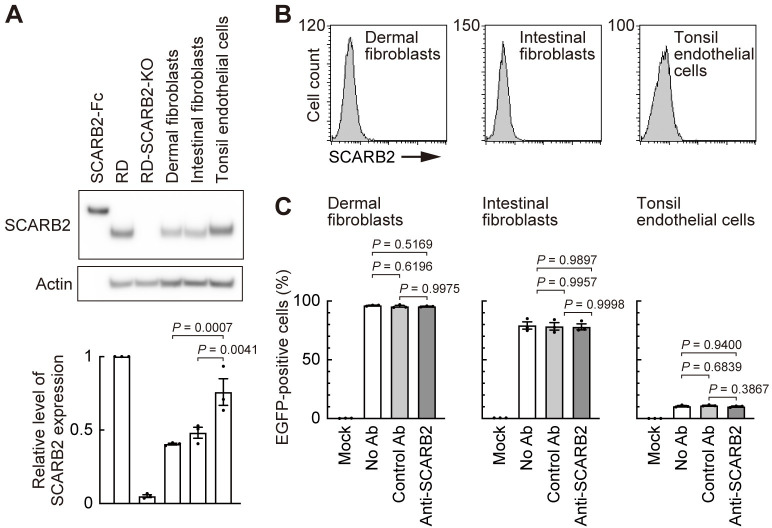
Enterovirus A71 (EV-A71) infection involves a variety of receptors. Among them, two transmembrane protein receptors have been investigated in detail and shown to be critical for infection: P-selectin glycoprotein ligand-1 (PSGL-1) in lymphocytes (Jurkat cells), and scavenger receptor class B member 2 (SCARB2) in rhabdomyosarcoma (RD) cells. PSGL-1 and SCARB2 have been reported to be expressed on the surface of Jurkat and RD cells, respectively. In the work reported here, we investigated the roles of PSGL-1 and SCARB2 in the process of EV-A71 entry. We first examined the expression of SCARB2 in Jurkat cells, and detected it within the cytoplasm, but not on the cell surface. Further, using PSGL-1 and SCARB2 knockout cells, we found that although both PSGL-1 and SCARB2 are essential for virus infection of Jurkat cells, virus attachment to these cells requires only PSGL-1. These results led us to evaluate the cell surface expression and the roles of SCARB2 in other EV-A71-susceptible cell lines. Surprisingly, in contrast to the results of previous studies, we found that SCARB2 is absent from the surface of RD cells and other susceptible cell lines we examined, and that although SCARB2 is essential for infection of these cells, it is dispensable for virus attachment. These results indicate that a receptor other than SCARB2 is responsible for virus attachment to the cell and probably for internalization of virions, not only in Jurkat cells but also in RD cells and other EV-A71-susceptible cells. SCARB2 is highly concentrated in lysosomes and late endosomes, where it is likely to trigger acid-dependent uncoating of virions, the critical final step of the entry process. Our results suggest that the essential interactions between EV-A71 and SCARB2 occur, not at the cell surface, but within the cell.
Copyright: © 2024 Nishimura et al. This is an open access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
