

Shared and distinct mechanisms of UBA1 inactivation across different diseases
- PMID: 38360993
- PMCID: PMC11099125
- DOI: 10.1038/s44318-024-00046-z
Shared and distinct mechanisms of UBA1 inactivation across different diseases
Abstract
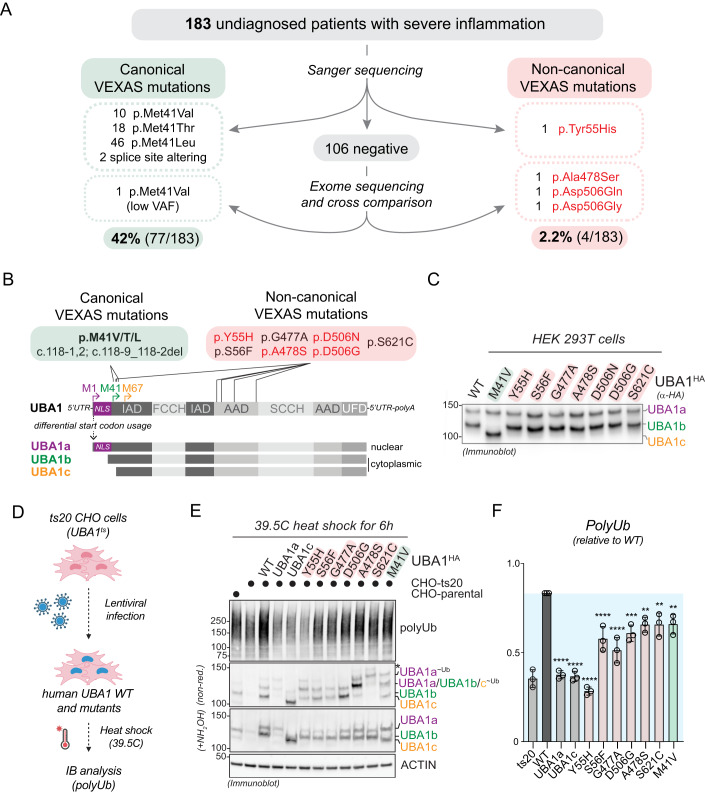
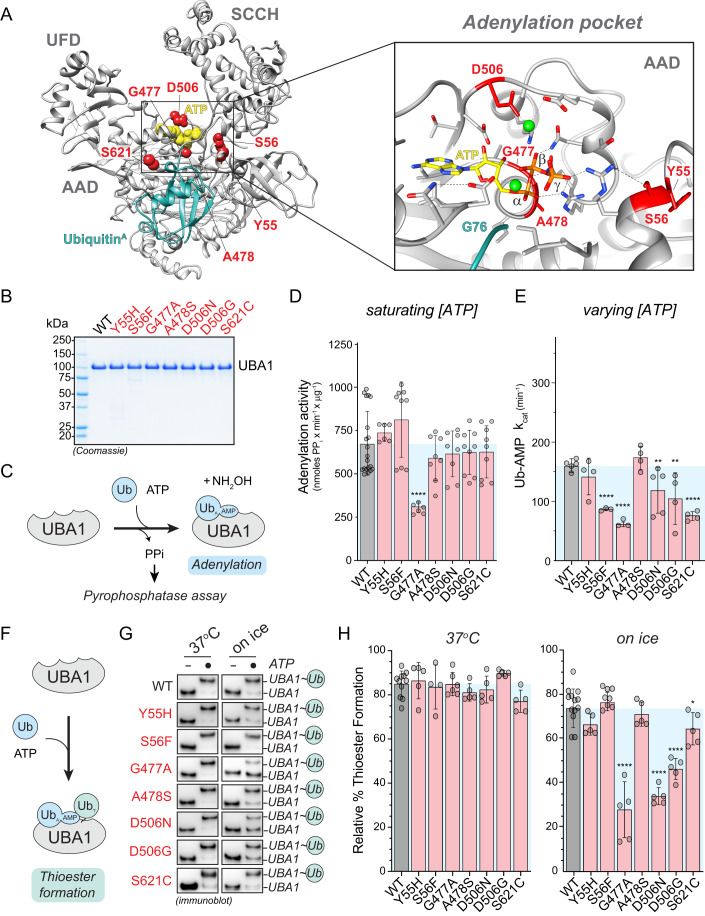
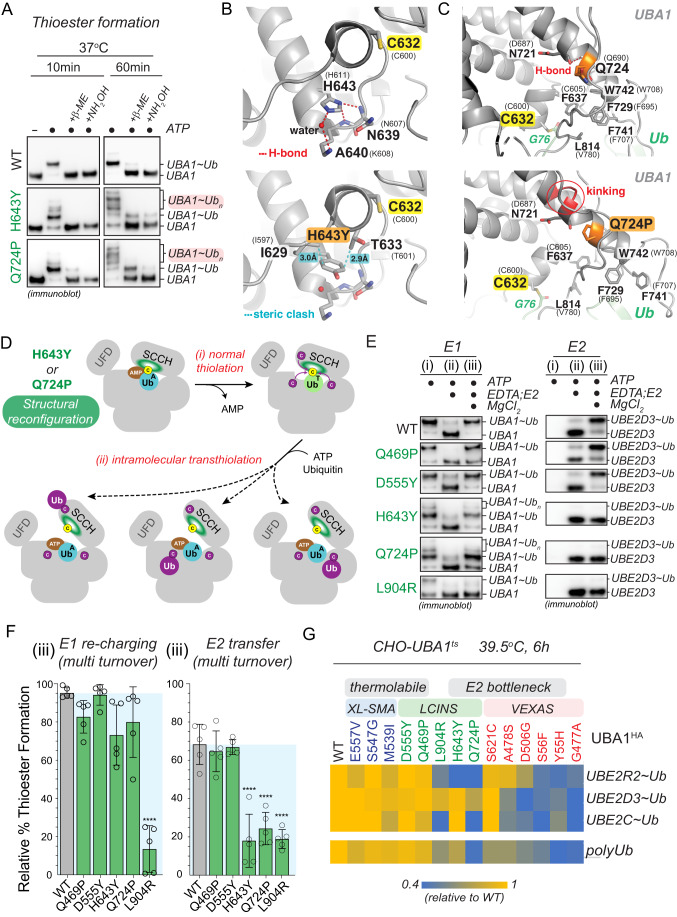
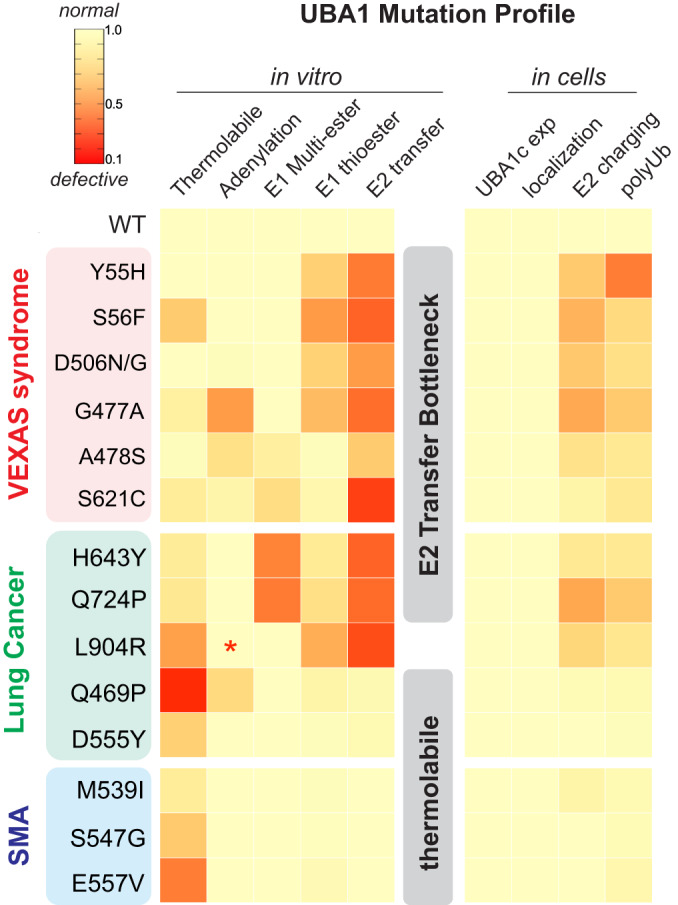
Most cellular ubiquitin signaling is initiated by UBA1, which activates and transfers ubiquitin to tens of E2 enzymes. Clonally acquired UBA1 missense mutations cause an inflammatory-hematologic overlap disease called VEXAS (vacuoles, E1, X-linked, autoinflammatory, somatic) syndrome. Despite extensive clinical investigation into this lethal disease, little is known about the underlying molecular mechanisms. Here, by dissecting VEXAS-causing UBA1 mutations, we discovered that p.Met41 mutations alter cytoplasmic isoform expression, whereas other mutations reduce catalytic activity of nuclear and cytoplasmic isoforms by diverse mechanisms, including aberrant oxyester formation. Strikingly, non-p.Met41 mutations most prominently affect transthioesterification, revealing ubiquitin transfer to cytoplasmic E2 enzymes as a shared property of pathogenesis amongst different VEXAS syndrome genotypes. A similar E2 charging bottleneck exists in some lung cancer-associated UBA1 mutations, but not in spinal muscular atrophy-causing UBA1 mutations, which instead, render UBA1 thermolabile. Collectively, our results highlight the precision of conformational changes required for faithful ubiquitin transfer, define distinct and shared mechanisms of UBA1 inactivation in diverse diseases, and suggest that specific E1-E2 modules control different aspects of tissue differentiation and maintenance.
Keywords: Lung Cancer in Never Smokers (LCINS); Spinal Muscular Atrophy (SMA); UBA1; Ubiquitin; VEXAS Syndrome.
© 2024. This is a U.S. Government work and not under copyright protection in the US; foreign copyright protection may apply.
Conflict of interest statement
The authors declare no competing interests.
Figures








Update of
-
Shared and Distinct Mechanisms of UBA1 Inactivation Across Different Diseases.bioRxiv [Preprint]. 2023 Oct 10:2023.10.10.561769. doi: 10.1101/2023.10.10.561769. bioRxiv. 2023. Update in: EMBO J. 2024 May;43(10):1919-1946. doi: 10.1038/s44318-024-00046-z. PMID: 37873213 Free PMC article. Updated. Preprint.
References
-
- Beck DB, Bodian DL, Shah V, Mirshahi UL, Kim J, Ding Y, Magaziner SJ, Strande NT, Cantor A, Haley JS, et al. Estimated prevalence and clinical manifestations of UBA1 variants associated with VEXAS syndrome in a clinical population. JAMA. 2023;329:318–324. doi: 10.1001/jama.2022.24836. - DOI - PMC - PubMed
MeSH terms
Substances
Grants and funding
- BMFRP- IDA HT9425-23-1-0507/U.S. Department of Defense (DOD)
- ZIC DE000744/ImNIH/Intramural NIH HHS/United States
- ZIA BC011490/ImNIH/Intramural NIH HHS/United States
- ZIA DE000749/ImNIH/Intramural NIH HHS/United States
- R00AR078205/HHS | NIH | National Institute of Arthritis and Musculoskeletal and Skin Diseases (NIAMS)
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
