

Drug-target affinity prediction with extended graph learning-convolutional networks
- PMID: 38365583
- PMCID: PMC10874073
- DOI: 10.1186/s12859-024-05698-6
Drug-target affinity prediction with extended graph learning-convolutional networks
Abstract
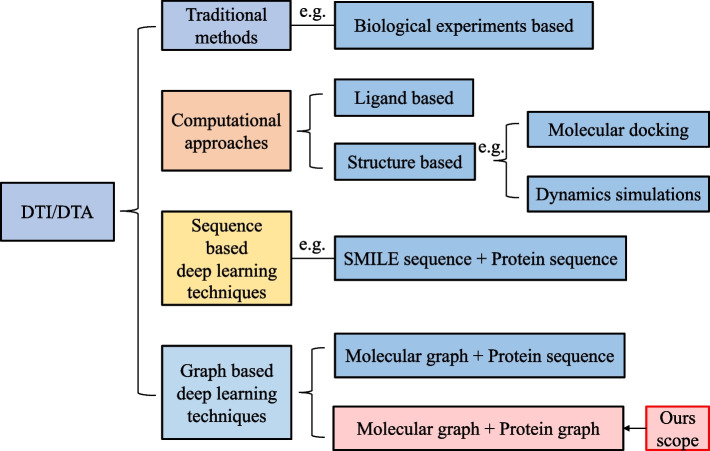
Background: High-performance computing plays a pivotal role in computer-aided drug design, a field that holds significant promise in pharmaceutical research. The prediction of drug-target affinity (DTA) is a crucial stage in this process, potentially accelerating drug development through rapid and extensive preliminary compound screening, while also minimizing resource utilization and costs. Recently, the incorporation of deep learning into DTA prediction and the enhancement of its accuracy have emerged as key areas of interest in the research community. Drugs and targets can be characterized through various methods, including structure-based, sequence-based, and graph-based representations. Despite the progress in structure and sequence-based techniques, they tend to provide limited feature information. Conversely, graph-based approaches have risen to prominence, attracting considerable attention for their comprehensive data representation capabilities. Recent studies have focused on constructing protein and drug molecular graphs using sequences and SMILES, subsequently deriving representations through graph neural networks. However, these graph-based approaches are limited by the use of a fixed adjacent matrix of protein and drug molecular graphs for graph convolution. This limitation restricts the learning of comprehensive feature representations from intricate compound and protein structures, consequently impeding the full potential of graph-based feature representation in DTA prediction. This, in turn, significantly impacts the models' generalization capabilities in the complex realm of drug discovery.
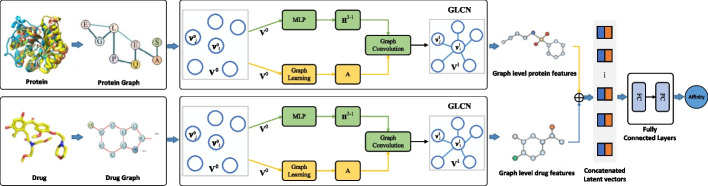
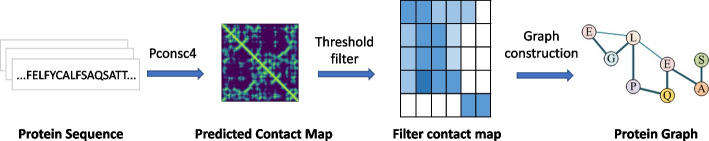
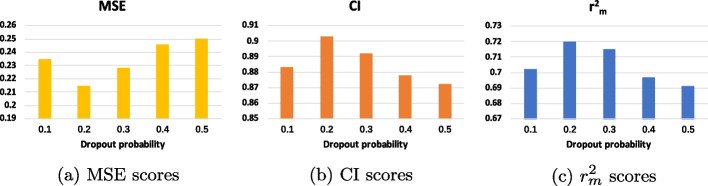
Results: To tackle these challenges, we introduce GLCN-DTA, a model specifically designed for proficiency in DTA tasks. GLCN-DTA innovatively integrates a graph learning module into the existing graph architecture. This module is designed to learn a soft adjacent matrix, which effectively and efficiently refines the contextual structure of protein and drug molecular graphs. This advancement allows for learning richer structural information from protein and drug molecular graphs via graph convolution, specifically tailored for DTA tasks, compared to the conventional fixed adjacent matrix approach. A series of experiments have been conducted to validate the efficacy of the proposed GLCN-DTA method across diverse scenarios. The results demonstrate that GLCN-DTA possesses advantages in terms of robustness and high accuracy.
Conclusions: The proposed GLCN-DTA model enhances DTA prediction performance by introducing a novel framework that synergizes graph learning operations with graph convolution operations, thereby achieving richer representations. GLCN-DTA does not distinguish between different protein classifications, including structurally ordered and intrinsically disordered proteins, focusing instead on improving feature representation. Therefore, its applicability scope may be more effective in scenarios involving structurally ordered proteins, while potentially being limited in contexts with intrinsically disordered proteins.
Keywords: Deep learning; Drug discovery; Drug–target affinity prediction; Graph learning-convolutional networks.
© 2024. The Author(s).
Conflict of interest statement
The authors declare that they have no competing interests.
Figures

Similar articles
-
GeneralizedDTA: combining pre-training and multi-task learning to predict drug-target binding affinity for unknown drug discovery.BMC Bioinformatics. 2022 Sep 7;23(1):367. doi: 10.1186/s12859-022-04905-6. BMC Bioinformatics. 2022. PMID: 36071406 Free PMC article.
-
A survey of drug-target interaction and affinity prediction methods via graph neural networks.Comput Biol Med. 2023 Sep;163:107136. doi: 10.1016/j.compbiomed.2023.107136. Epub 2023 Jun 7. Comput Biol Med. 2023. PMID: 37329615 Review.
-
MSGNN-DTA: Multi-Scale Topological Feature Fusion Based on Graph Neural Networks for Drug-Target Binding Affinity Prediction.Int J Mol Sci. 2023 May 5;24(9):8326. doi: 10.3390/ijms24098326. Int J Mol Sci. 2023. PMID: 37176031 Free PMC article.
-
G-K BertDTA: A graph representation learning and semantic embedding-based framework for drug-target affinity prediction.Comput Biol Med. 2024 May;173:108376. doi: 10.1016/j.compbiomed.2024.108376. Epub 2024 Mar 25. Comput Biol Med. 2024. PMID: 38552281
-
A comprehensive review of the recent advances on predicting drug-target affinity based on deep learning.Front Pharmacol. 2024 Apr 2;15:1375522. doi: 10.3389/fphar.2024.1375522. eCollection 2024. Front Pharmacol. 2024. PMID: 38628639 Free PMC article. Review.
Cited by
-
Attention-based approach to predict drug-target interactions across seven target superfamilies.Bioinformatics. 2024 Aug 2;40(8):btae496. doi: 10.1093/bioinformatics/btae496. Bioinformatics. 2024. PMID: 39115379 Free PMC article.
-
A Multibranch Neural Network for Drug-Target Affinity Prediction Using Similarity Information.ACS Omega. 2024 Aug 12;9(33):35978-35989. doi: 10.1021/acsomega.4c05607. eCollection 2024 Aug 20. ACS Omega. 2024. PMID: 39184467 Free PMC article.
-
DTBA-net: Drug-Target Binding Affinity prediction using feature selection in hybrid CNN model.J Comput Aided Mol Des. 2025 Jun 16;39(1):31. doi: 10.1007/s10822-025-00605-4. J Comput Aided Mol Des. 2025. PMID: 40522546
-
Advances in Protein-Ligand Binding Affinity Prediction via Deep Learning: A Comprehensive Study of Datasets, Data Preprocessing Techniques, and Model Architectures.Curr Drug Targets. 2024;25(15):1041-1065. doi: 10.2174/0113894501330963240905083020. Curr Drug Targets. 2024. PMID: 39318214 Free PMC article. Review.
-
Drug target affinity prediction based on multi-scale gated power graph and multi-head linear attention mechanism.PLoS One. 2025 Feb 21;20(2):e0315718. doi: 10.1371/journal.pone.0315718. eCollection 2025. PLoS One. 2025. PMID: 39982887 Free PMC article.
References
-
- Mullard A. New drugs cost us$2.6 billion to develop. Nat Rev Drug Discov. 2014;13:877–877.
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
