

Optimized bacterial community characterization through full-length 16S rRNA gene sequencing utilizing MinION nanopore technology
- PMID: 38365589
- PMCID: PMC10870487
- DOI: 10.1186/s12866-024-03208-5
Optimized bacterial community characterization through full-length 16S rRNA gene sequencing utilizing MinION nanopore technology
Abstract
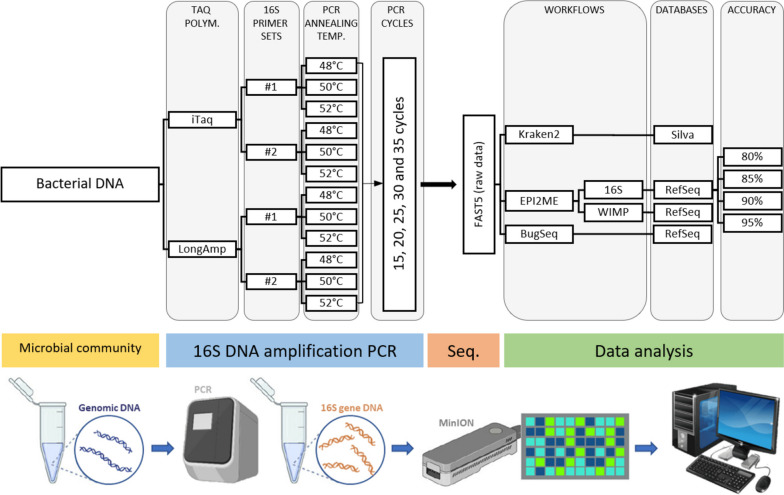
Background: Accurate identification of bacterial communities is crucial for research applications, diagnostics, and clinical interventions. Although 16S ribosomal RNA (rRNA) gene sequencing is a widely employed technique for bacterial taxonomic classification, it often results in misclassified or unclassified bacterial taxa. This study sought to refine the full-length 16S rRNA gene sequencing protocol using the MinION sequencer, focusing on the V1-V9 regions. Our methodological enquiry examined several factors, including the number of PCR amplification cycles, choice of primers and Taq polymerase, and specific sequence databases and workflows employed. We used a microbial standard comprising eight bacterial strains (five gram-positive and three gram-negative) in known proportions as a validation control.
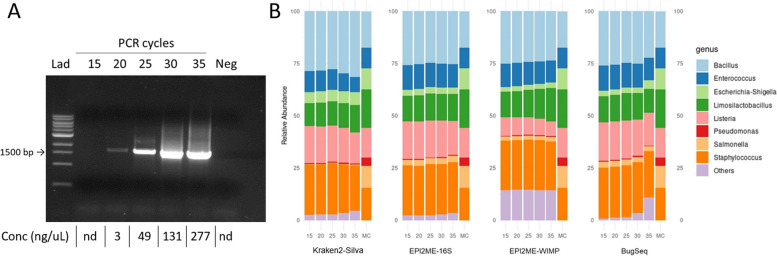
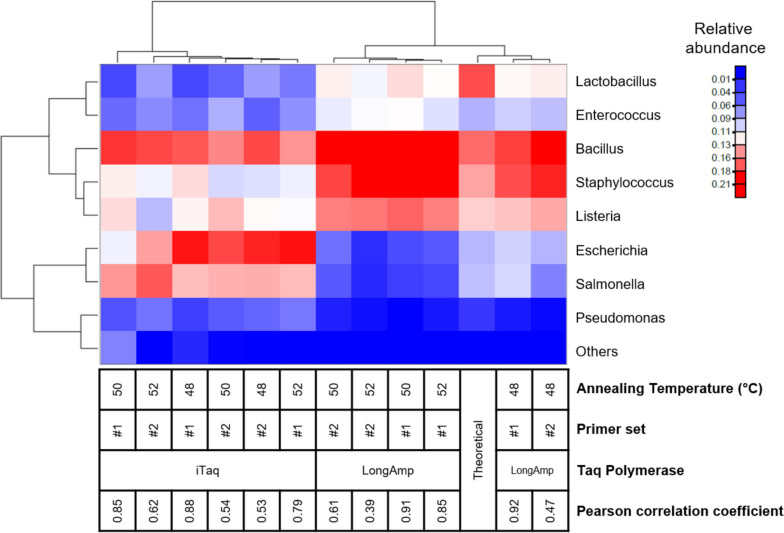
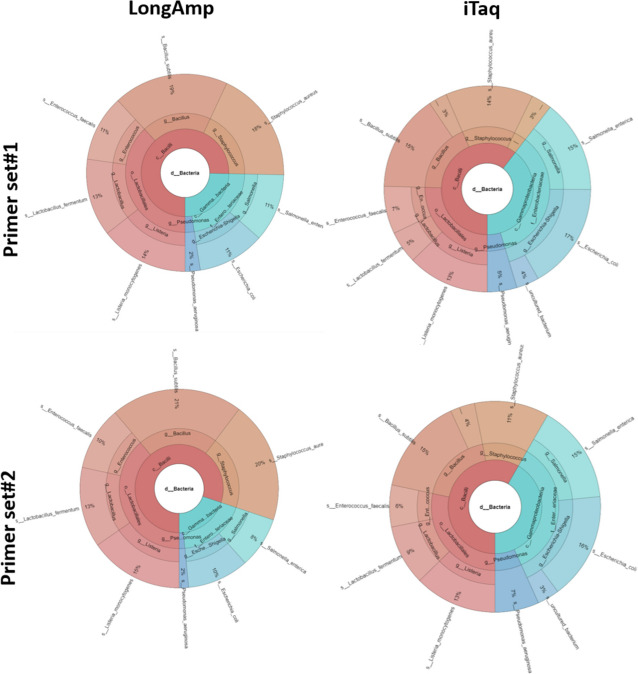
Results: Based on the MinION protocol, we employed the microbial standard as the DNA template for the 16S rRNA gene amplicon sequencing procedure. Our analysis showed that an elevated number of PCR amplification cycles introduced PCR bias, and the selection of Taq polymerase and primer sets significantly affected the subsequent analysis. Bacterial identification at genus level demonstrated Pearson correlation coefficients ranging from 0.73 to 0.79 when assessed using BugSeq, Kraken-Silva and EPI2ME-16S workflows. Notably, the EPI2ME-16S workflow exhibited the highest Pearson correlation with the microbial standard, minimised misclassification, and increased alignment accuracy. At the species taxonomic level, the BugSeq workflow was superior, with a Pearson correlation coefficient of 0.92.
Conclusions: These findings emphasise the importance of careful selection of PCR settings and a well-structured analytical framework for 16S rRNA full-length gene sequencing. The results showed a robust correlation between the predicted and observed bacterial abundances at both the genus and species taxonomic levels, making these findings applicable across diverse research contexts and with clinical utility for reliable pathogen identification.
Keywords: 16S rRNA gene-based sequencing; Bacterial DNA; Nanopore sequencing; V1–V9 region.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
Similar articles
-
Comparison of Illumina versus Nanopore 16S rRNA Gene Sequencing of the Human Nasal Microbiota.Genes (Basel). 2020 Sep 21;11(9):1105. doi: 10.3390/genes11091105. Genes (Basel). 2020. PMID: 32967250 Free PMC article.
-
Species-level resolution of 16S rRNA gene amplicons sequenced through the MinION™ portable nanopore sequencer.Gigascience. 2016 Jan 28;5:4. doi: 10.1186/s13742-016-0111-z. eCollection 2016. Gigascience. 2016. PMID: 26823973 Free PMC article.
-
MinION™ nanopore sequencing of environmental metagenomes: a synthetic approach.Gigascience. 2017 Mar 1;6(3):1-10. doi: 10.1093/gigascience/gix007. Gigascience. 2017. PMID: 28327976 Free PMC article.
-
Research Techniques Made Simple: Bacterial 16S Ribosomal RNA Gene Sequencing in Cutaneous Research.J Invest Dermatol. 2016 Mar;136(3):e23-e27. doi: 10.1016/j.jid.2016.01.005. J Invest Dermatol. 2016. PMID: 26902128 Free PMC article. Review.
-
16S, 18S gene, and ITS region Sequencing: Expanded Applications in Pathology Diagnostics and Research.Cesk Patol. 2025;60(4):176-180. Cesk Patol. 2025. PMID: 40088441 Review. English.
Cited by
-
Microbiomes of frozen blood plasma samples reveal potential pathogens in wild birds and rodents.One Health. 2025 Jun 11;21:101109. doi: 10.1016/j.onehlt.2025.101109. eCollection 2025 Dec. One Health. 2025. PMID: 40606019 Free PMC article.
-
Investigating the Microbial Dynamics of Hermetia illucens Powder Throughout Rearing and Processing: An Integrated Approach Using Cultural and Metabarcoding Methods.Foods. 2025 Jun 20;14(13):2161. doi: 10.3390/foods14132161. Foods. 2025. PMID: 40646912 Free PMC article.
-
16S rDNA Sequencing for Bacterial Identification in Preterm Infants with Suspected Early-Onset Neonatal Sepsis.Trop Med Infect Dis. 2024 Jul 6;9(7):152. doi: 10.3390/tropicalmed9070152. Trop Med Infect Dis. 2024. PMID: 39058194 Free PMC article.
-
16S rRNA phylogeny and clustering is not a reliable proxy for genome-based taxonomy in Streptomyces.Microb Genom. 2024 Sep;10(9):001287. doi: 10.1099/mgen.0.001287. Microb Genom. 2024. PMID: 39254673 Free PMC article.
-
Antibiotic resistance patterns of environmental bacteria from sewage water in Vellore, India: isolation, virulence analysis, and characterization.Front Microbiol. 2025 Aug 12;16:1640369. doi: 10.3389/fmicb.2025.1640369. eCollection 2025. Front Microbiol. 2025. PMID: 40873707 Free PMC article.
