

The cell biology of ferroptosis
- PMID: 38366038
- PMCID: PMC12187608
- DOI: 10.1038/s41580-024-00703-5
The cell biology of ferroptosis
Abstract
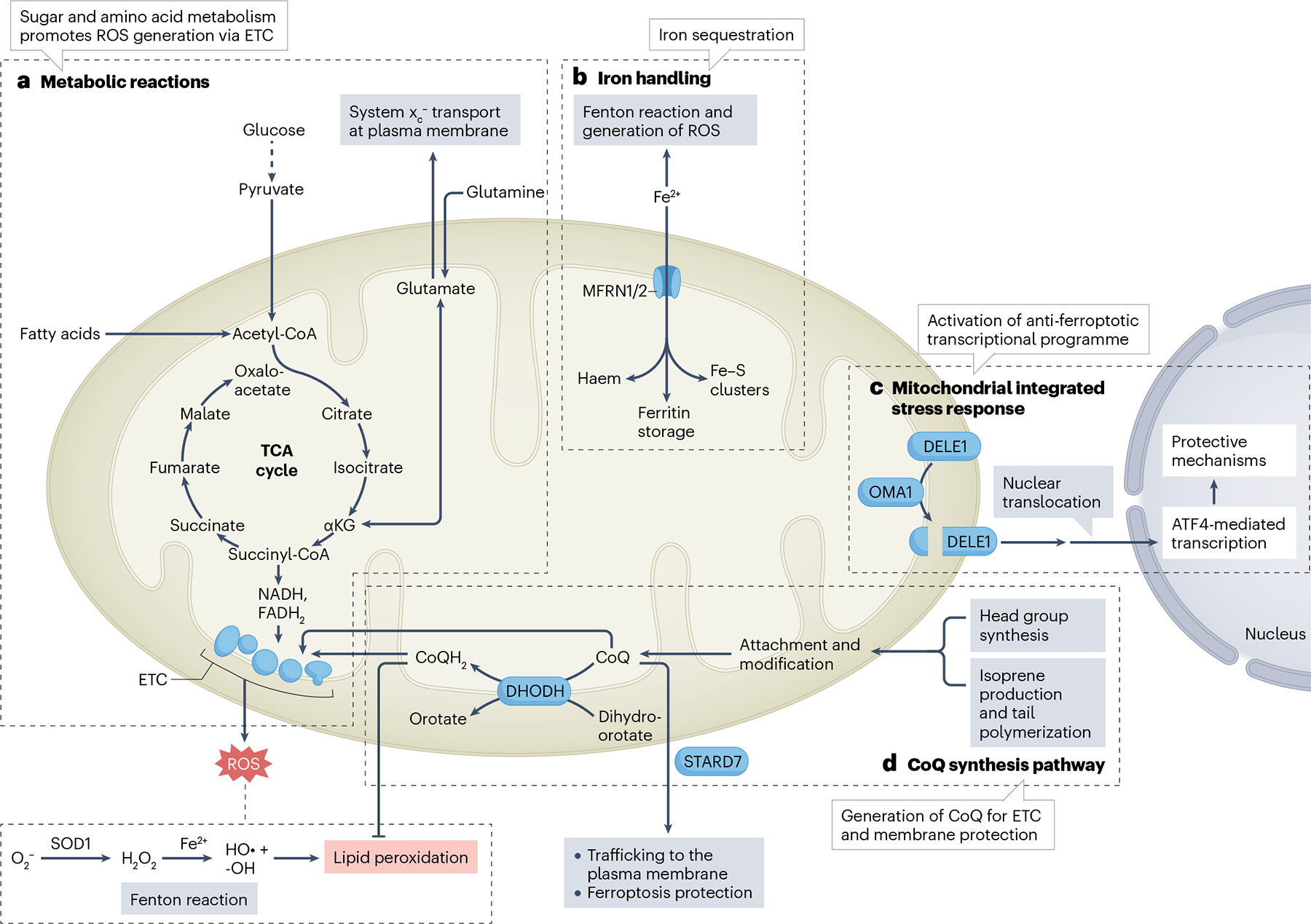
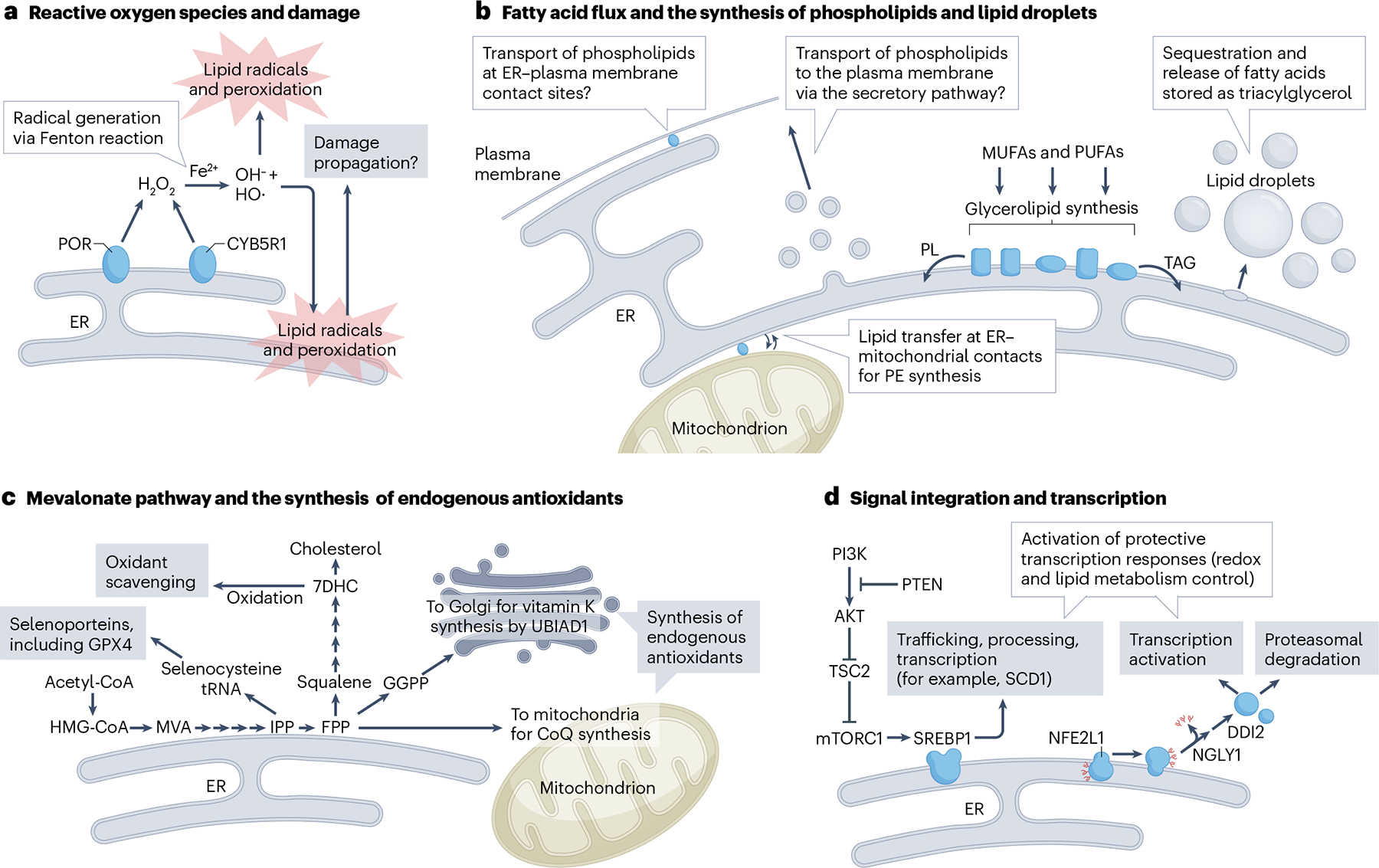
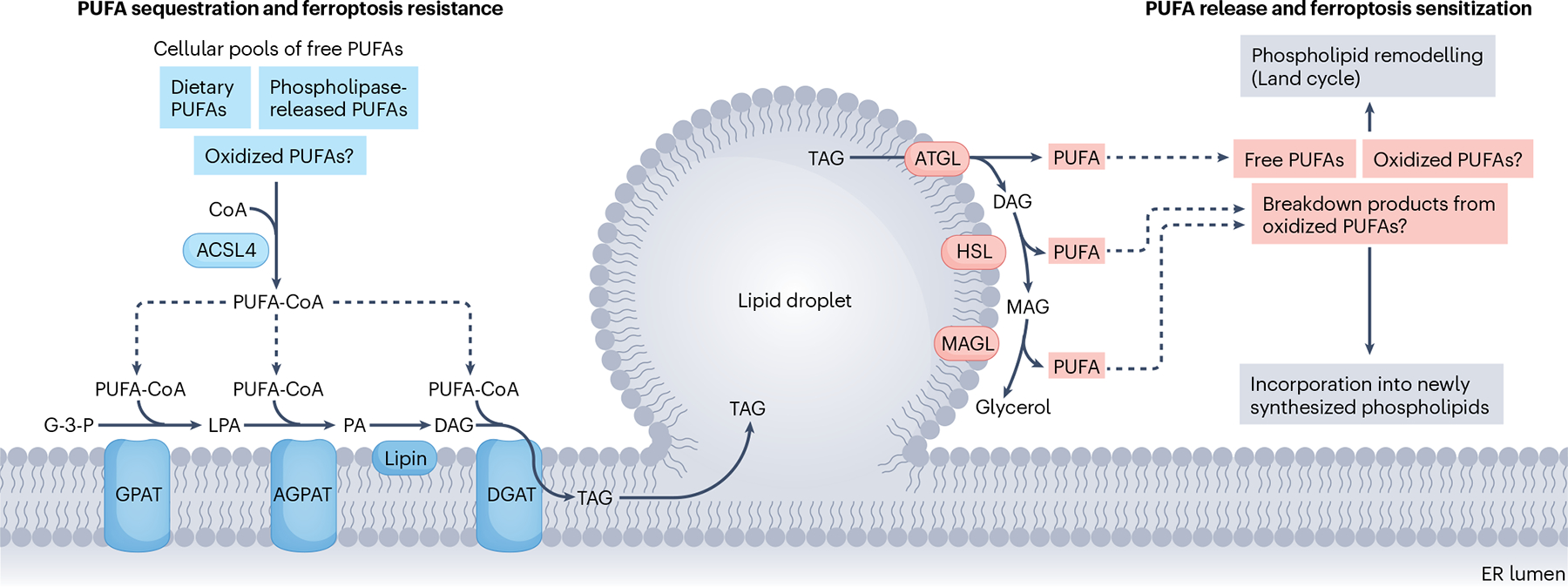
Ferroptosis is a non-apoptotic cell death mechanism characterized by iron-dependent membrane lipid peroxidation. Here, we review what is known about the cellular mechanisms mediating the execution and regulation of ferroptosis. We first consider how the accumulation of membrane lipid peroxides leads to the execution of ferroptosis by altering ion transport across the plasma membrane. We then discuss how metabolites and enzymes that are distributed in different compartments and organelles throughout the cell can regulate sensitivity to ferroptosis by impinging upon iron, lipid and redox metabolism. Indeed, metabolic pathways that reside in the mitochondria, endoplasmic reticulum, lipid droplets, peroxisomes and other organelles all contribute to the regulation of ferroptosis sensitivity. We note how the regulation of ferroptosis sensitivity by these different organelles and pathways seems to vary between different cells and death-inducing conditions. We also highlight transcriptional master regulators that integrate the functions of different pathways and organelles to modulate ferroptosis sensitivity globally. Throughout this Review, we highlight open questions and areas in which progress is needed to better understand the cell biology of ferroptosis.
© 2024. Springer Nature Limited.
Conflict of interest statement
Competing interests
S.J.D. is a co-founder of Prothegen and a member of the scientific advisory board for Hillstream BioPharma. S.J.D. holds patents related to ferroptosis. J.A.O. is a member of the scientific advisory board for Vicinitas Therapeutics and holds patents related to ferroptosis.
Figures


References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
