

Alternative polyadenylation quantitative trait methylation mapping in human cancers provides clues into the molecular mechanisms of APA
- PMID: 38367620
- PMCID: PMC10940021
- DOI: 10.1016/j.ajhg.2024.01.010
Alternative polyadenylation quantitative trait methylation mapping in human cancers provides clues into the molecular mechanisms of APA
Abstract
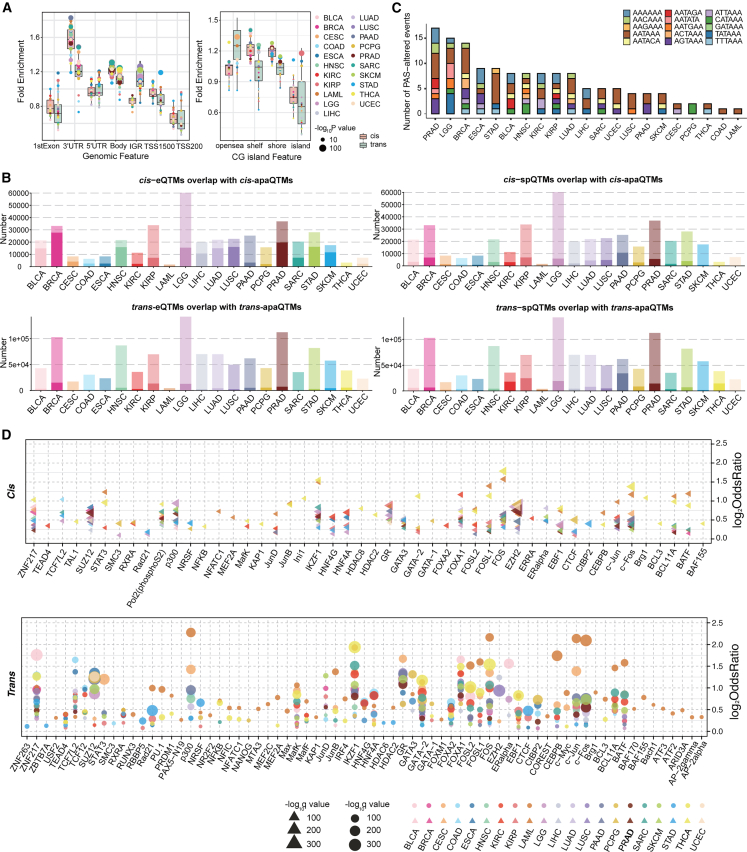
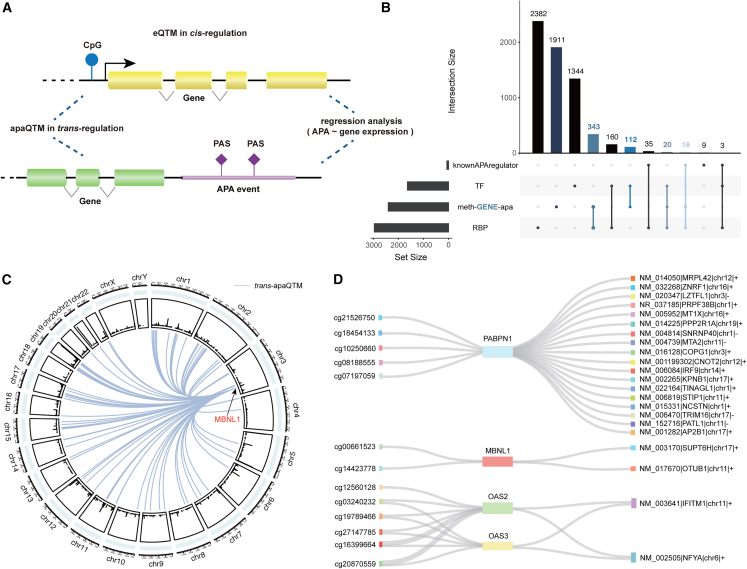
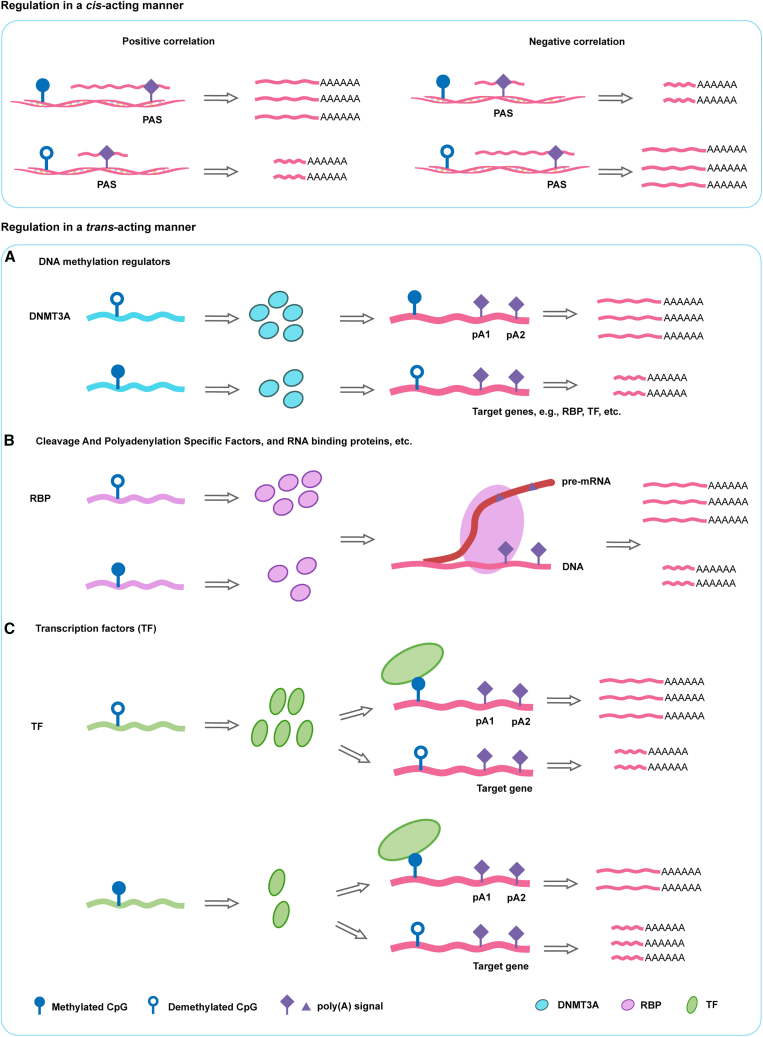
Genetic variants are involved in the orchestration of alternative polyadenylation (APA) events, while the role of DNA methylation in regulating APA remains unclear. We generated a comprehensive atlas of APA quantitative trait methylation sites (apaQTMs) across 21 different types of cancer (1,612 to 60,219 acting in cis and 4,448 to 142,349 in trans). Potential causal apaQTMs in non-cancer samples were also identified. Mechanistically, we observed a strong enrichment of cis-apaQTMs near polyadenylation sites (PASs) and both cis- and trans-apaQTMs in proximity to transcription factor (TF) binding regions. Through the integration of ChIP-signals and RNA-seq data from cell lines, we have identified several regulators of APA events, acting either directly or indirectly, implicating novel functions of some important genes, such as TCF7L2, which is known for its involvement in type 2 diabetes and cancers. Furthermore, we have identified a vast number of QTMs that share the same putative causal CpG sites with five different cancer types, underscoring the roles of QTMs, including apaQTMs, in the process of tumorigenesis. DNA methylation is extensively involved in the regulation of APA events in human cancers. In an attempt to elucidate the potential underlying molecular mechanisms of APA by DNA methylation, our study paves the way for subsequent experimental validations into the intricate biological functions of DNA methylation in APA regulation and the pathogenesis of human cancers. To present a comprehensive catalog of apaQTM patterns, we introduce the Pancan-apaQTM database, available at https://pancan-apaqtm-zju.shinyapps.io/pancanaQTM/.
Keywords: DNA methylation; Mendelian randomization analysis; alternative polyadenylation; and colocalization; cis-regulation; human cancer; pan-cancer analysis; post-transcription; quantitative trait methylation sites; trans-regulation.
Copyright © 2024 American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests The authors declare no competing interests.
Figures





Similar articles
-
Genetic Variants That Impact Alternative Polyadenylation in Cancer Represent Candidate Causal Risk Loci.Cancer Res. 2023 Nov 1;83(21):3650-3666. doi: 10.1158/0008-5472.CAN-23-0251. Cancer Res. 2023. PMID: 37669142
-
Colocalization analysis of 3' UTR alternative polyadenylation quantitative trait loci reveals novel mechanisms underlying associations with lung function.Hum Mol Genet. 2024 Jun 21;33(13):1164-1175. doi: 10.1093/hmg/ddae055. Hum Mol Genet. 2024. PMID: 38569558 Free PMC article.
-
Recurrent tumor-specific regulation of alternative polyadenylation of cancer-related genes.BMC Genomics. 2018 Jul 13;19(1):536. doi: 10.1186/s12864-018-4903-7. BMC Genomics. 2018. PMID: 30005633 Free PMC article.
-
Advances of functional consequences and regulation mechanisms of alternative cleavage and polyadenylation.Yi Chuan. 2021 Jan 20;43(1):4-15. doi: 10.16288/j.yczz.20-200. Yi Chuan. 2021. PMID: 33509770 Review.
-
RNA-binding proteins in regulation of alternative cleavage and polyadenylation.Adv Exp Med Biol. 2014;825:97-127. doi: 10.1007/978-1-4939-1221-6_3. Adv Exp Med Biol. 2014. PMID: 25201104 Review.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
