

Mucosal host-microbe interactions associate with clinical phenotypes in inflammatory bowel disease
- PMID: 38368394
- PMCID: PMC10874382
- DOI: 10.1038/s41467-024-45855-2
Mucosal host-microbe interactions associate with clinical phenotypes in inflammatory bowel disease
Abstract
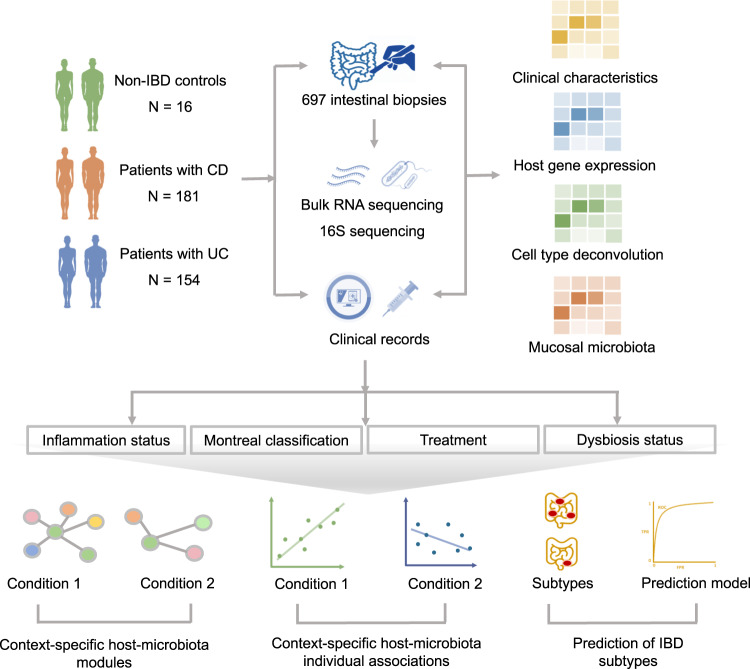
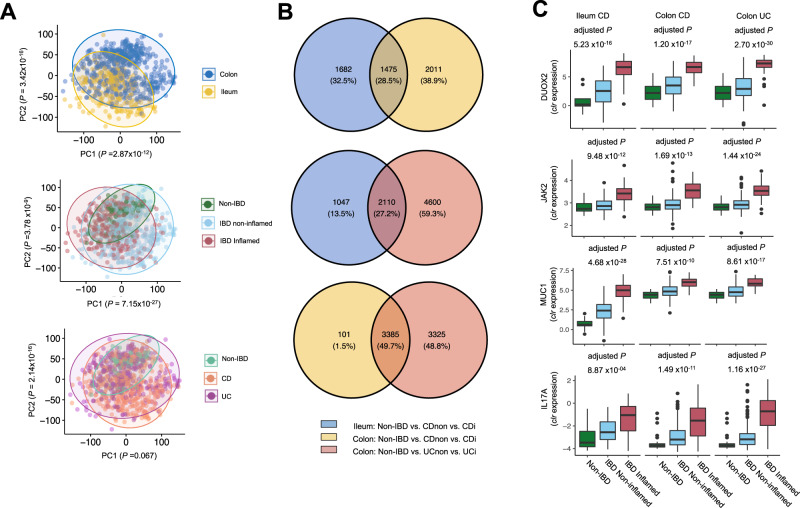
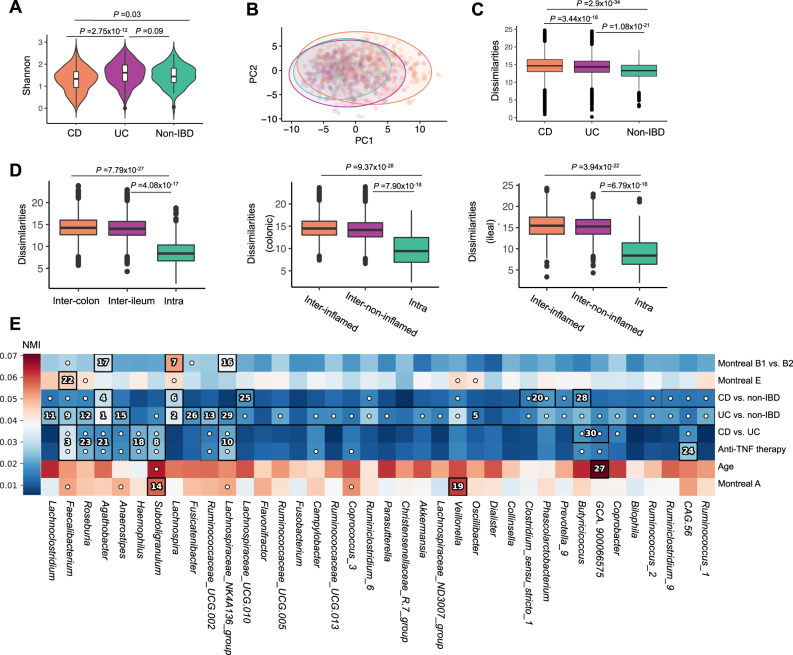
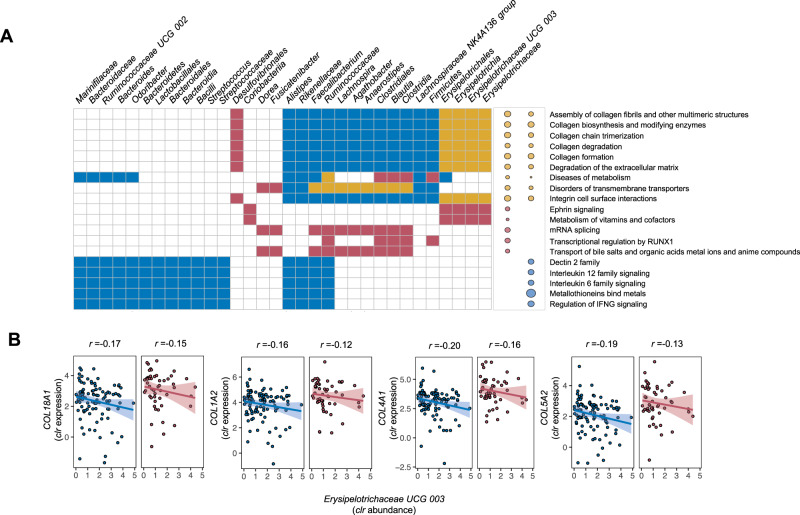
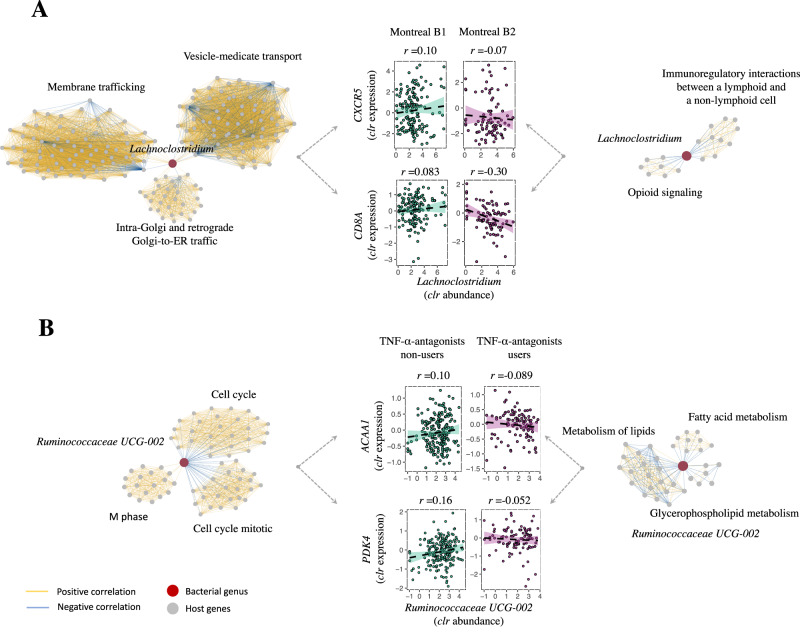
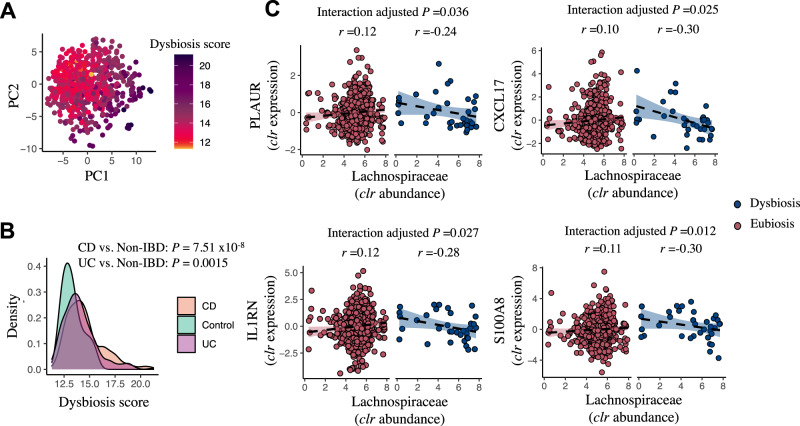
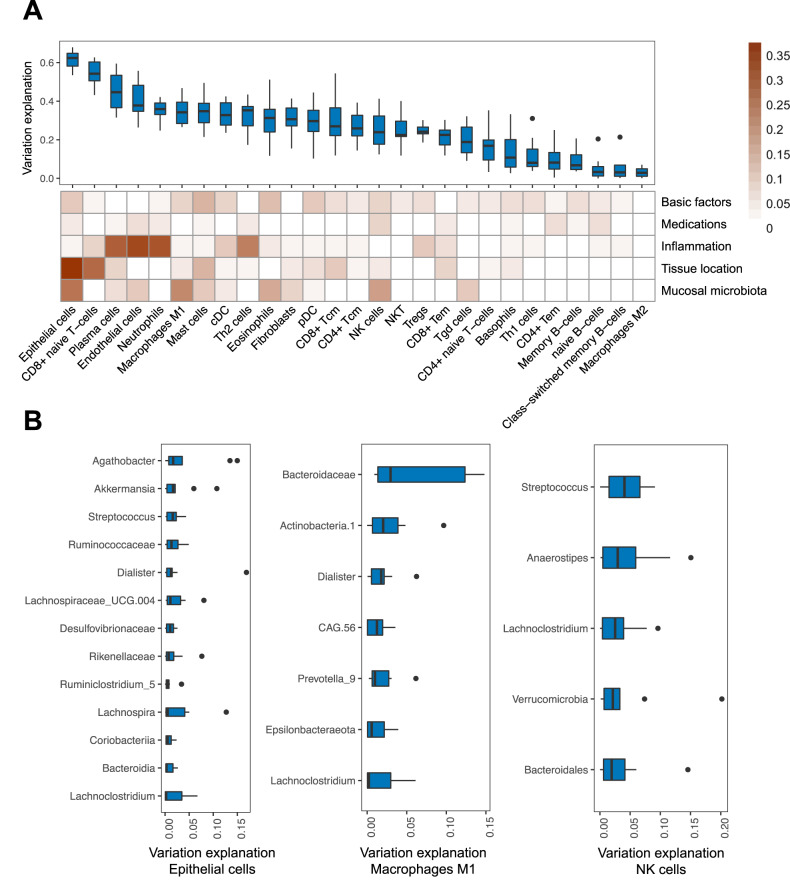
Disrupted host-microbe interactions at the mucosal level are key to the pathophysiology of IBD. This study aimed to comprehensively examine crosstalk between mucosal gene expression and microbiota in patients with IBD. To study tissue-specific interactions, we perform transcriptomic (RNA-seq) and microbial (16S-rRNA-seq) profiling of 697 intestinal biopsies (645 derived from 335 patients with IBD and 52 from 16 non-IBD controls). Mucosal gene expression patterns in IBD are mainly determined by tissue location and inflammation, whereas the mucosal microbiota composition shows a high degree of individual specificity. Analysis of transcript-bacteria interactions identifies six distinct groups of inflammation-related pathways that are associated with intestinal microbiota (adjusted P < 0.05). An increased abundance of Bifidobacterium is associated with higher expression of genes involved in fatty acid metabolism, while Bacteroides correlates with increased metallothionein signaling. In patients with fibrostenosis, a transcriptional network dominated by immunoregulatory genes is associated with Lachnoclostridium bacteria in non-stenotic tissue (adjusted P < 0.05), while being absent in CD without fibrostenosis. In patients using TNF-α-antagonists, a transcriptional network dominated by fatty acid metabolism genes is linked to Ruminococcaceae (adjusted P < 0.05). Mucosal microbiota composition correlates with enrichment of intestinal epithelial cells, macrophages, and NK-cells. Overall, these data demonstrate the presence of context-specific mucosal host-microbe interactions in IBD, revealing significantly altered inflammation-associated gene-taxa modules, particularly in patients with fibrostenotic CD and patients using TNF-α-antagonists. This study provides compelling insights into host-microbe interactions that may guide microbiota-directed precision medicine and fuels the rationale for microbiota-targeted therapeutics as a strategy to alter disease course in IBD.
© 2024. The Author(s).
Conflict of interest statement
RKW acted as a consultant for Takeda, received unrestricted research grants from Takeda, Johnson & Johnson, Tramedico and Ferring and received speaker fees from MSD, Abbvie, and Janssen Pharmaceuticals. GD received an unrestricted research grant from Takeda and speaker fees from Pfizer and Janssen Pharmaceuticals. All other authors declare no competing interests.
Figures
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
