

Assessing the efficacy of target adaptive sampling long-read sequencing through hereditary cancer patient genomes
- PMID: 38368425
- PMCID: PMC10874402
- DOI: 10.1038/s41525-024-00394-z
Assessing the efficacy of target adaptive sampling long-read sequencing through hereditary cancer patient genomes
Abstract
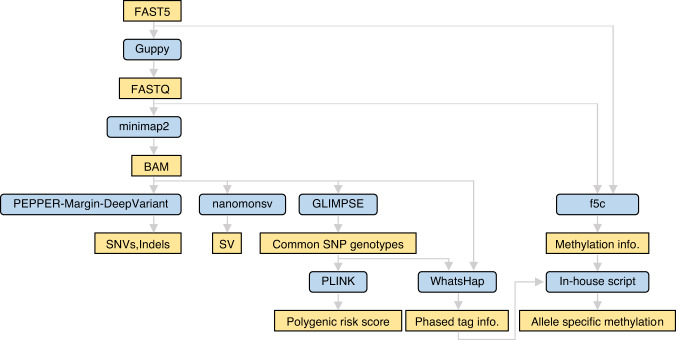
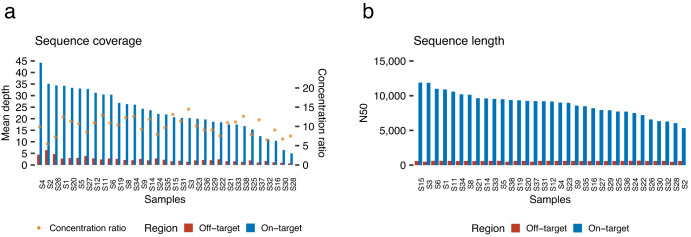
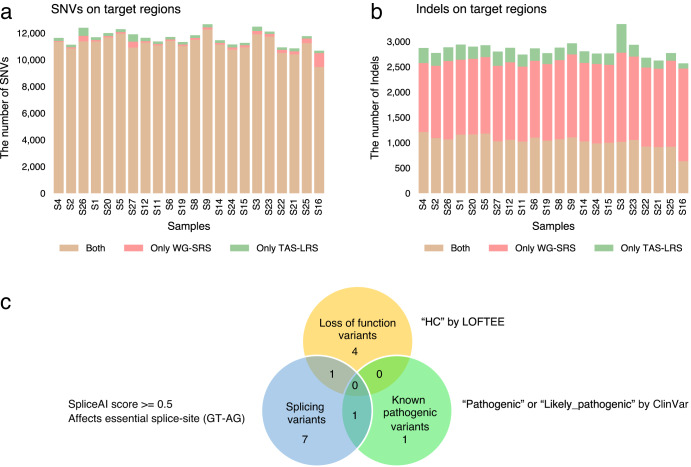
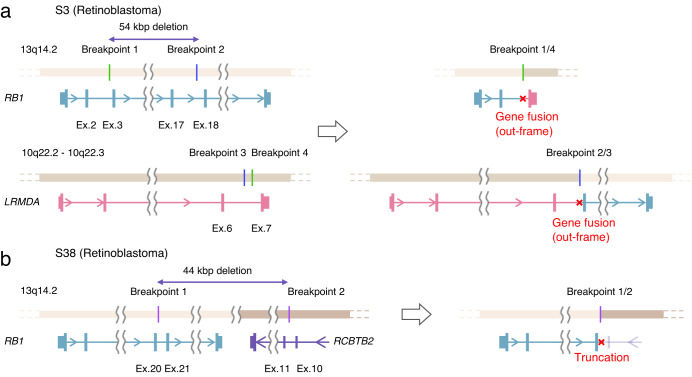
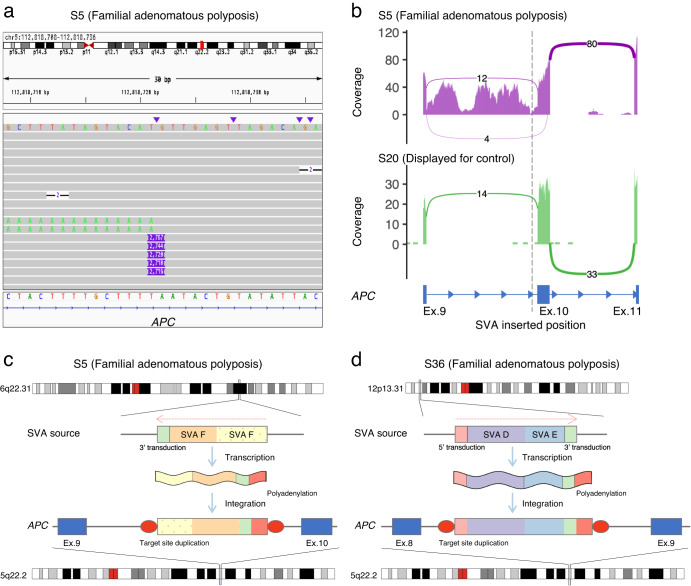
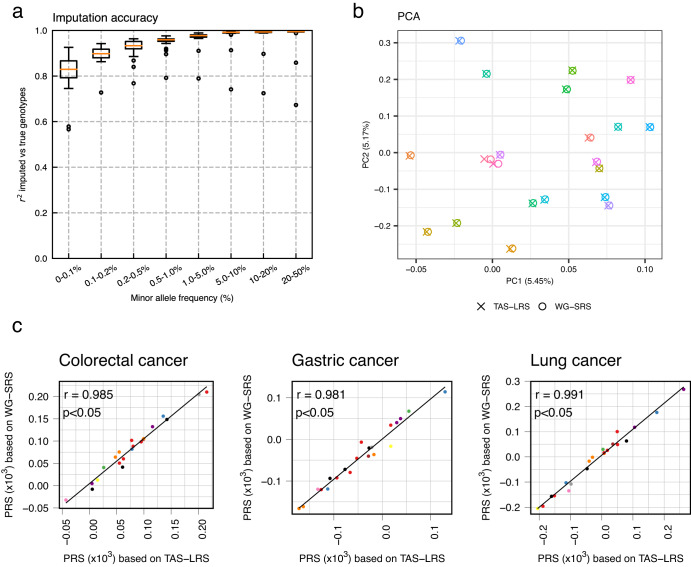
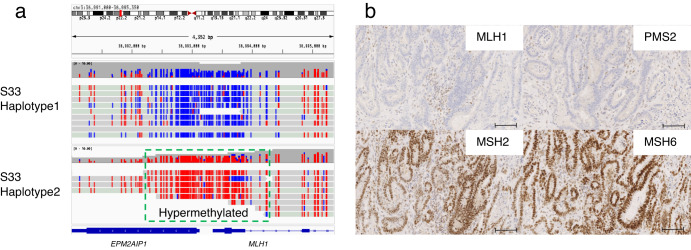
Innovations in sequencing technology have led to the discovery of novel mutations that cause inherited diseases. However, many patients with suspected genetic diseases remain undiagnosed. Long-read sequencing technologies are expected to significantly improve the diagnostic rate by overcoming the limitations of short-read sequencing. In addition, Oxford Nanopore Technologies (ONT) offers adaptive sampling and computationally driven target enrichment technology. This enables more affordable intensive analysis of target gene regions compared to standard non-selective long-read sequencing. In this study, we developed an efficient computational workflow for target adaptive sampling long-read sequencing (TAS-LRS) and evaluated it through application to 33 genomes collected from suspected hereditary cancer patients. Our workflow can identify single nucleotide variants with nearly the same accuracy as the short-read platform and elucidate complex forms of structural variations. We also newly identified several SINE-R/VNTR/Alu (SVA) elements affecting the APC gene in two patients with familial adenomatous polyposis, as well as their sites of origin. In addition, we demonstrated that off-target reads from adaptive sampling, which is typically discarded, can be effectively used to accurately genotype common single-nucleotide polymorphisms (SNPs) across the entire genome, enabling the calculation of a polygenic risk score. Furthermore, we identified allele-specific MLH1 promoter hypermethylation in a Lynch syndrome patient. In summary, our workflow with TAS-LRS can simultaneously capture monogenic risk variants including complex structural variations, polygenic background as well as epigenetic alterations, and will be an efficient platform for genetic disease research and diagnosis.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Grants and funding
LinkOut - more resources
Full Text Sources
