

This is a preprint.
Spatially Exploring RNA Biology in Archival Formalin-Fixed Paraffin-Embedded Tissues
- PMID: 38370833
- PMCID: PMC10871202
- DOI: 10.1101/2024.02.06.579143
Spatially Exploring RNA Biology in Archival Formalin-Fixed Paraffin-Embedded Tissues
Update in
-
Spatially exploring RNA biology in archival formalin-fixed paraffin-embedded tissues.Cell. 2024 Nov 14;187(23):6760-6779.e24. doi: 10.1016/j.cell.2024.09.001. Epub 2024 Sep 30. Cell. 2024. PMID: 39353436
Abstract
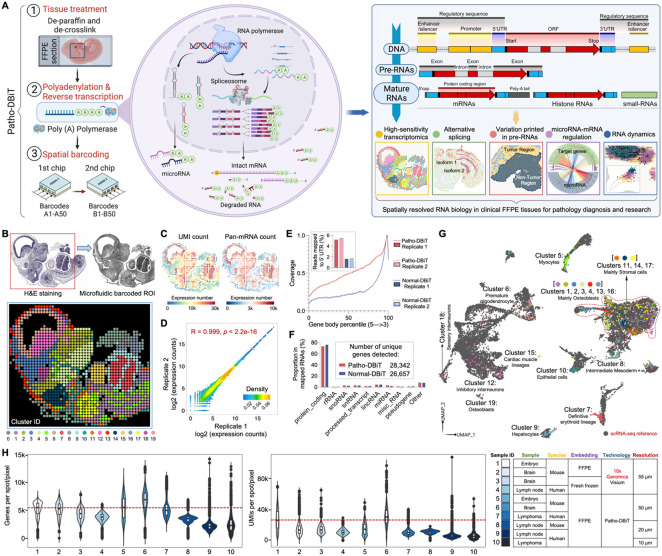
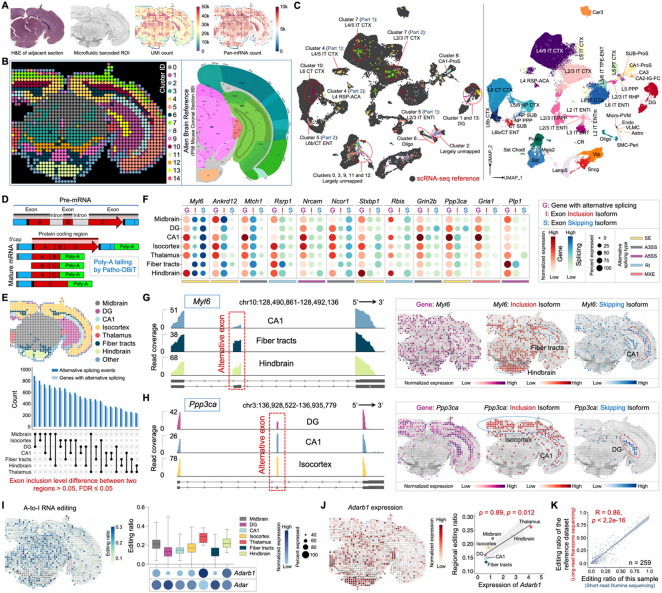
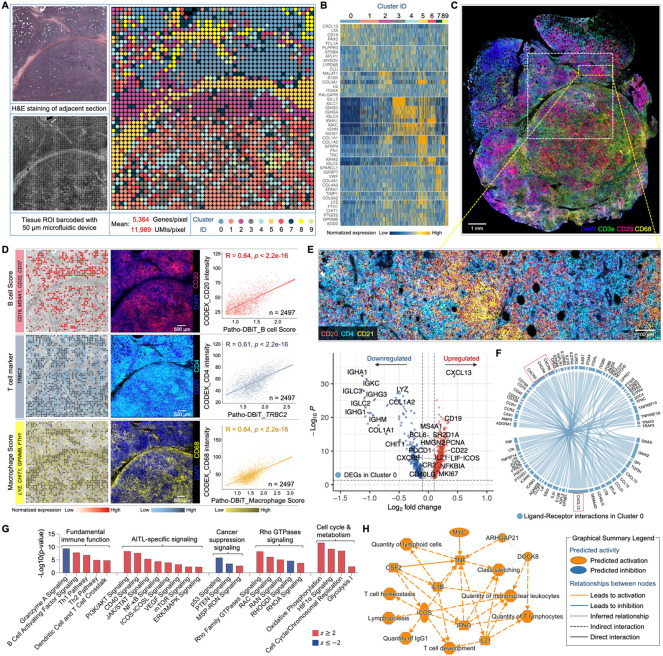
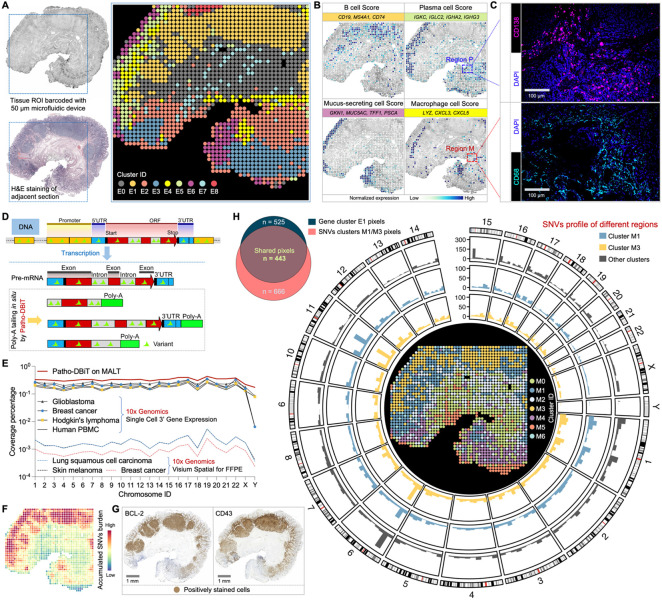
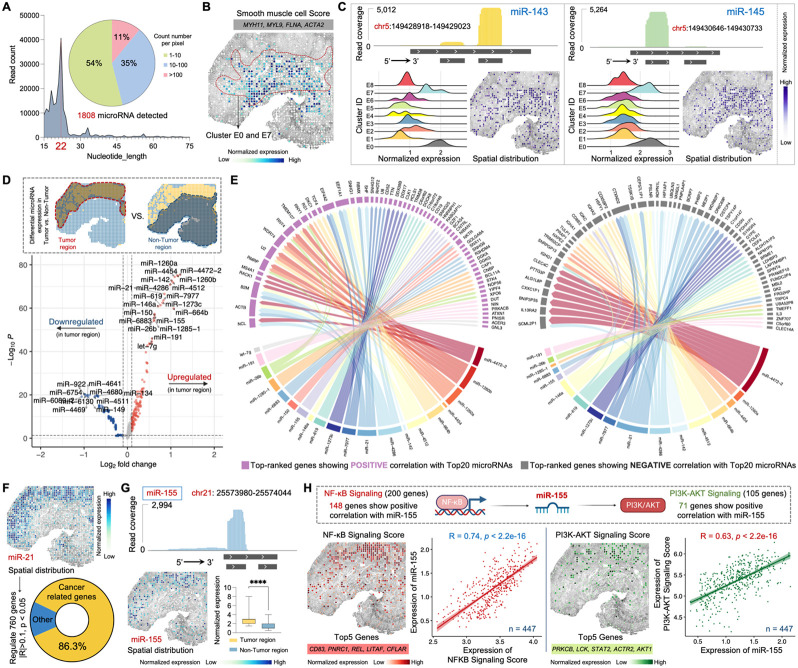
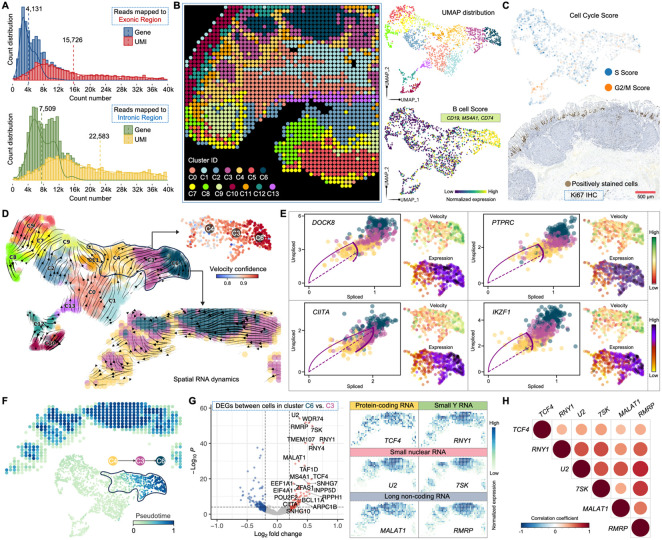
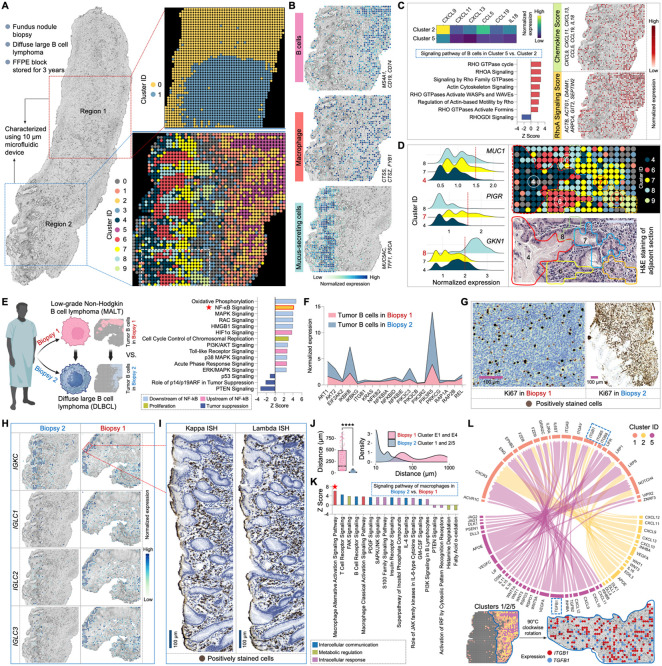
Spatial transcriptomics has emerged as a powerful tool for dissecting spatial cellular heterogeneity but as of today is largely limited to gene expression analysis. Yet, the life of RNA molecules is multifaceted and dynamic, requiring spatial profiling of different RNA species throughout the life cycle to delve into the intricate RNA biology in complex tissues. Human disease-relevant tissues are commonly preserved as formalin-fixed and paraffin-embedded (FFPE) blocks, representing an important resource for human tissue specimens. The capability to spatially explore RNA biology in FFPE tissues holds transformative potential for human biology research and clinical histopathology. Here, we present Patho-DBiT combining in situ polyadenylation and deterministic barcoding for spatial full coverage transcriptome sequencing, tailored for probing the diverse landscape of RNA species even in clinically archived FFPE samples. It permits spatial co-profiling of gene expression and RNA processing, unveiling region-specific splicing isoforms, and high-sensitivity transcriptomic mapping of clinical tumor FFPE tissues stored for five years. Furthermore, genome-wide single nucleotide RNA variants can be captured to distinguish different malignant clones from non-malignant cells in human lymphomas. Patho-DBiT also maps microRNA-mRNA regulatory networks and RNA splicing dynamics, decoding their roles in spatial tumorigenesis trajectory. High resolution Patho-DBiT at the cellular level reveals a spatial neighborhood and traces the spatiotemporal kinetics driving tumor progression. Patho-DBiT stands poised as a valuable platform to unravel rich RNA biology in FFPE tissues to study human tissue biology and aid in clinical pathology evaluation.
Conflict of interest statement
Declaration of Interest Z.B. and R.F. are inventors of a patent application related to this work. R.F. is scientific founder and adviser for IsoPlexis, Singleron Biotechnologies, and AtlasXomics. The interests of R.F. were reviewed and managed by Yale University Provost’s Office in accordance with the University’s conflict of interest policies. M.L.X. has served as consultant for Treeline Biosciences, Pure Marrow, and Seattle Genetics. Other authors declare no competing interests.
Figures
References
Publication types
Grants and funding
- R01 GM138856/GM/NIGMS NIH HHS/United States
- R56 HG012310/HG/NHGRI NIH HHS/United States
- R33 CA246711/CA/NCI NIH HHS/United States
- U54 CA274509/CA/NCI NIH HHS/United States
- RF1 MH128876/MH/NIMH NIH HHS/United States
- U54 CA268083/CA/NCI NIH HHS/United States
- UH3 CA257393/CA/NCI NIH HHS/United States
- U54 DK106857/DK/NIDDK NIH HHS/United States
- R01 CA245313/CA/NCI NIH HHS/United States
- U54 AG079759/AG/NIA NIH HHS/United States
- UM1 MH130991/MH/NIMH NIH HHS/United States
- U54 AG076043/AG/NIA NIH HHS/United States
- RM1 MH132648/MH/NIMH NIH HHS/United States
LinkOut - more resources
Full Text Sources