

TREX reveals proteins that bind to specific RNA regions in living cells
- PMID: 38374261
- PMCID: PMC10927567
- DOI: 10.1038/s41592-024-02181-1
TREX reveals proteins that bind to specific RNA regions in living cells
Abstract
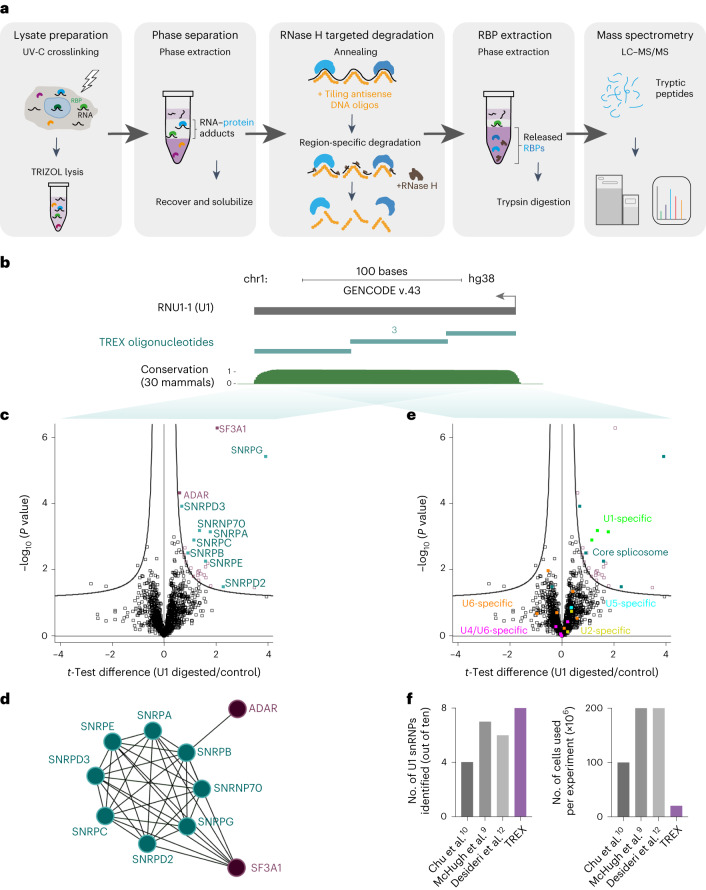
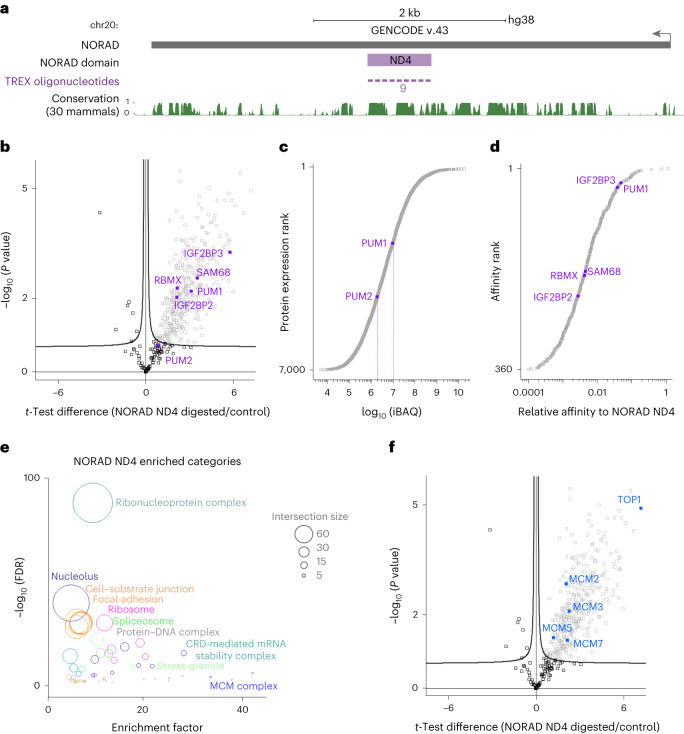
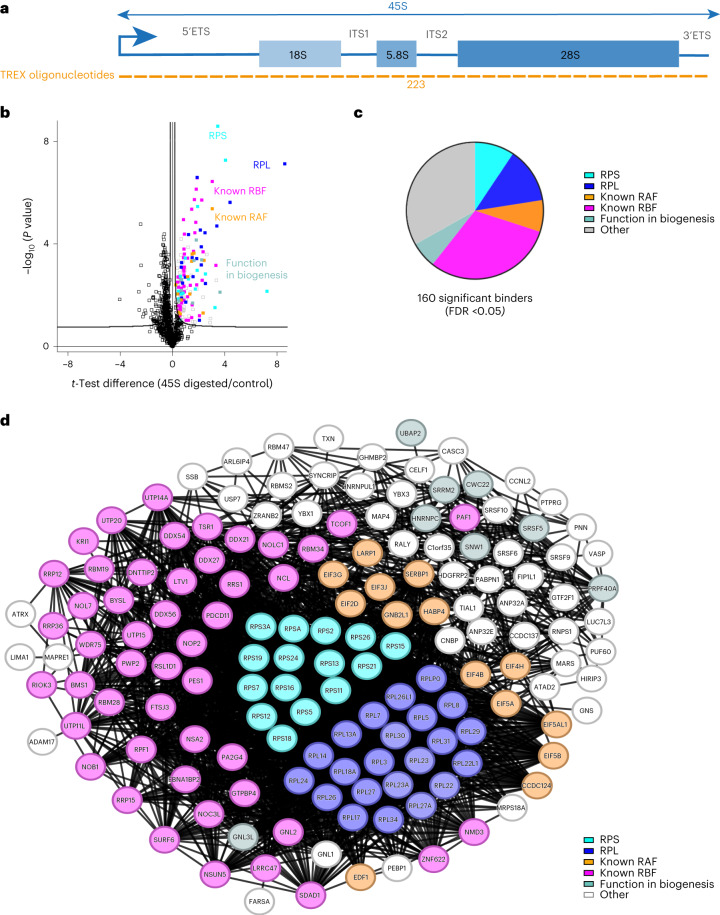
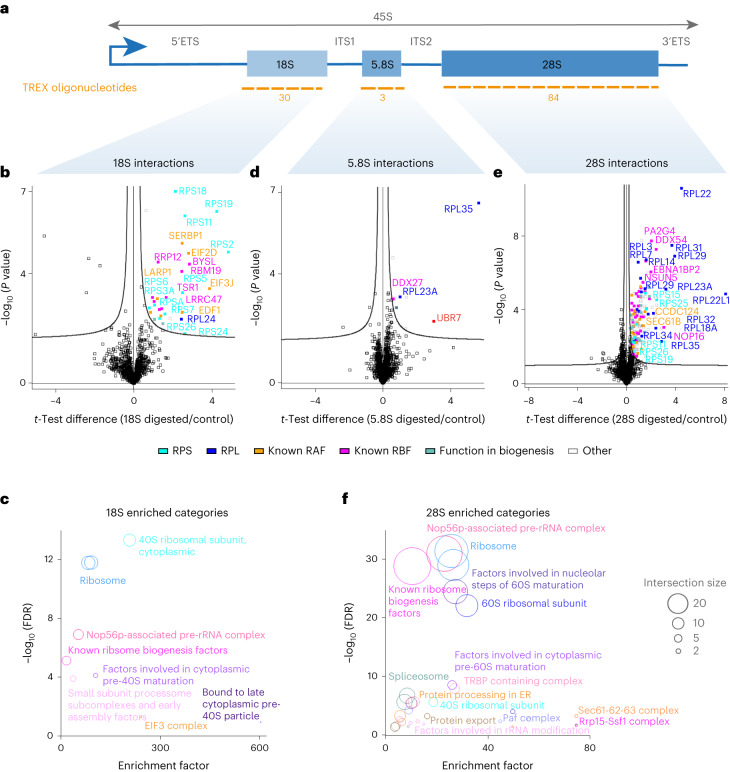
Different regions of RNA molecules can often engage in specific interactions with distinct RNA-binding proteins (RBPs), giving rise to diverse modalities of RNA regulation and function. However, there are currently no methods for unbiased identification of RBPs that interact with specific RNA regions in living cells and under endogenous settings. Here we introduce TREX (targeted RNase H-mediated extraction of crosslinked RBPs)-a highly sensitive approach for identifying proteins that directly bind to specific RNA regions in living cells. We demonstrate that TREX outperforms existing methods in identifying known interactors of U1 snRNA, and reveals endogenous region-specific interactors of NORAD long noncoding RNA. Using TREX, we generated a comprehensive region-by-region interactome for 45S rRNA, uncovering both established and previously unknown interactions that regulate ribosome biogenesis. With its applicability to different cell types, TREX is an RNA-centric tool for unbiased positional mapping of endogenous RNA-protein interactions in living cells.
© 2024. The Author(s).
Conflict of interest statement
F.K.M., L.S., M. Dermit, G.G. and M. Dodel are inventors and contributors on a pending patent covering the TREX method. The other authors declare no competing interests.
Figures








References
-
- Hafner M, et al. CLIP and complementary methods. Nat. Rev. Methods Prim. 2021;1:20. doi: 10.1038/s43586-021-00018-1. - DOI
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
