

Virus sequencing performance during the SARS-CoV-2 pandemic: a retrospective analysis of data from multiple rounds of external quality assessment in Austria
- PMID: 38375507
- PMCID: PMC10875003
- DOI: 10.3389/fmolb.2024.1327699
Virus sequencing performance during the SARS-CoV-2 pandemic: a retrospective analysis of data from multiple rounds of external quality assessment in Austria
Abstract
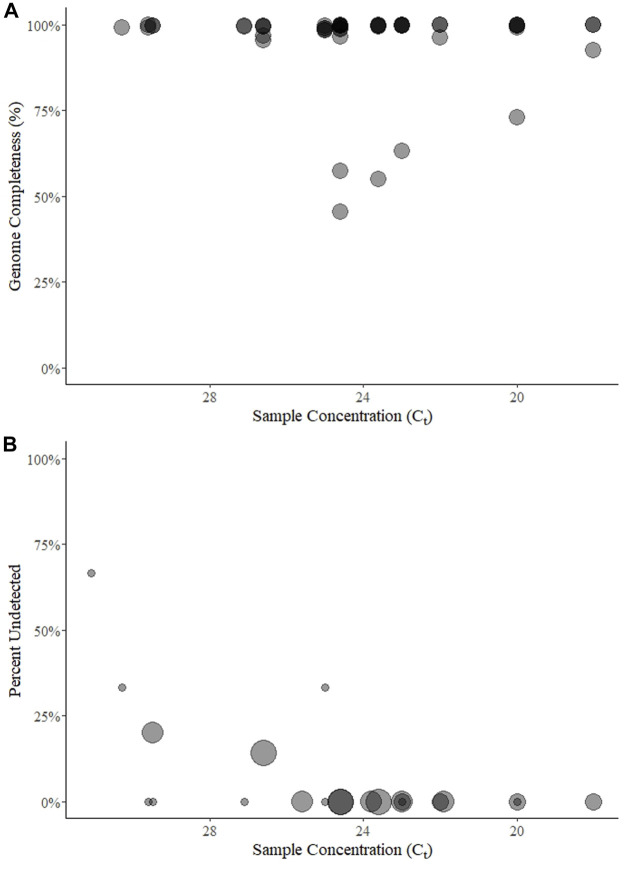
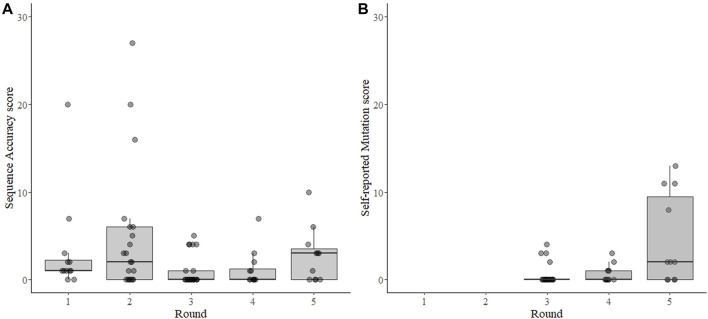
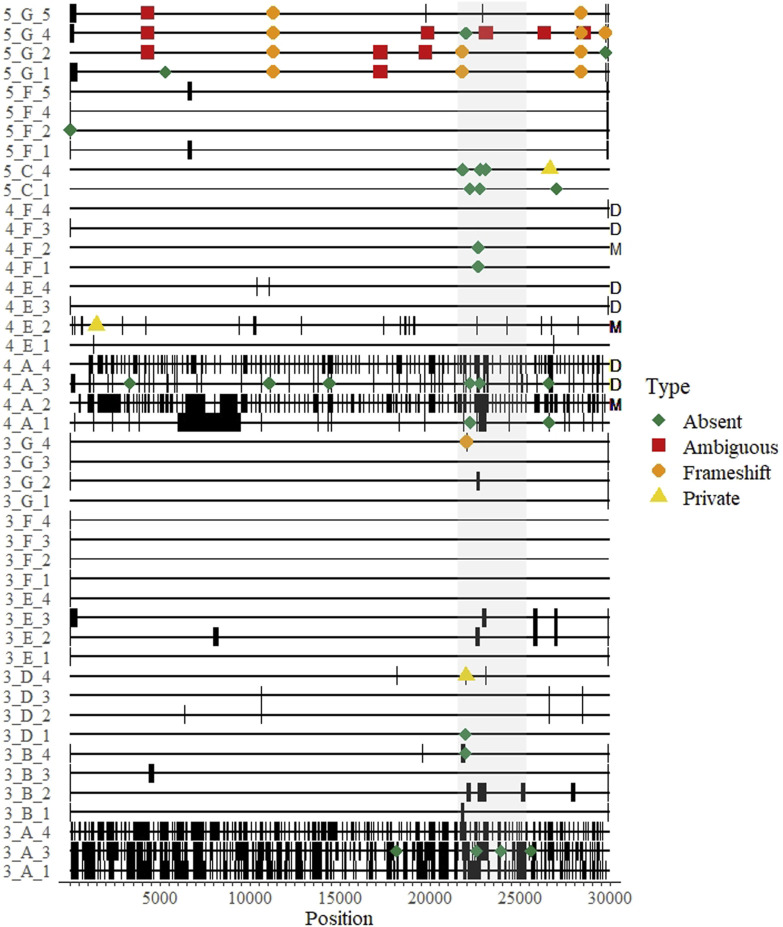
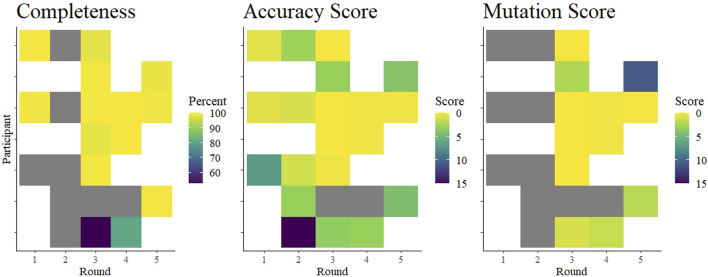
Introduction: A notable feature of the 2019 coronavirus disease (COVID-19) pandemic was the widespread use of whole genome sequencing (WGS) to monitor severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infections. Countries around the world relied on sequencing and other forms of variant detection to perform contact tracing and monitor changes in the virus genome, in the hopes that epidemic waves caused by variants would be detected and managed earlier. As sequencing was encouraged and rewarded by the government in Austria, but represented a new technicque for many laboratories, we designed an external quality assessment (EQA) scheme to monitor the accuracy of WGS and assist laboratories in validating their methods. Methods: We implemented SARS-CoV-2 WGS EQAs in Austria and report the results from 7 participants over 5 rounds from February 2021 until June 2023. The participants received sample material, sequenced genomes with routine methods, and provided the sequences as well as information about mutations and lineages. Participants were evaluated on the completeness and accuracy of the submitted sequence and the ability to analyze and interpret sequencing data. Results: The results indicate that performance was excellent with few exceptions, and these exceptions showed improvement over time. We extend our findings to infer that most publicly available sequences are accurate within ≤1 nucleotide, somewhat randomly distributed through the genome. Conclusion: WGS continues to be used for SARS-CoV-2 surveillance, and will likely be instrumental in future outbreak scenarios. We identified hurdles in building next-generation sequencing capacity in diagnostic laboratories. EQAs will help individual laboratories maintain high quality next-generation sequencing output, and strengthen variant monitoring and molecular epidemiology efforts.
Keywords: Austria; COVID-19 diagnostic testing; SARS-CoV-2; diagnostic laboratories; next-generation sequencing.
Copyright © 2024 Camp, Puchhammer-Stöckl, Aberle and Buchta.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
Similar articles
-
Performance of SARS-CoV-2 nucleic acid amplification testing in Austria as measured by external quality assessment schemes during 3 years of the COVID-19 pandemic: an observational retrospective study.Lancet Microbe. 2023 Dec;4(12):e1015-e1023. doi: 10.1016/S2666-5247(23)00286-0. Epub 2023 Nov 15. Lancet Microbe. 2023. PMID: 37979591
-
External quality assessment of molecular detection and variant typing of SARS-CoV-2 in European expert laboratories in 2023.J Clin Microbiol. 2025 Apr 9;63(4):e0153824. doi: 10.1128/jcm.01538-24. Epub 2025 Mar 14. J Clin Microbiol. 2025. PMID: 40084838 Free PMC article.
-
External quality assessments for SARS-CoV-2 genome detection in Austria : A comparison of the first postpandemic round with results from the pandemic era.Wien Klin Wochenschr. 2024 Aug;136(15-16):429-438. doi: 10.1007/s00508-024-02353-1. Epub 2024 Apr 23. Wien Klin Wochenschr. 2024. PMID: 38653873 Free PMC article.
-
Continued improvement in the development of the SARS-CoV-2 whole genome sequencing proficiency testing program.Pathology. 2024 Aug;56(5):717-725. doi: 10.1016/j.pathol.2024.02.010. Epub 2024 Apr 24. Pathology. 2024. PMID: 38729860
-
Molecular biology of SARS-CoV-2 and techniques of diagnosis and surveillance.Adv Clin Chem. 2024;118:35-85. doi: 10.1016/bs.acc.2023.11.003. Epub 2023 Dec 14. Adv Clin Chem. 2024. PMID: 38280807 Review.
References
-
- Angers-Loustau A., Petrillo M., Bengtsson-Palme J., Berendonk T., Blais B., Chan K. G., et al. (2018). The challenges of designing a benchmark strategy for bioinformatics pipelines in the identification of antimicrobial resistance determinants using next generation sequencing technologies. F1000Res. F1000Res, ISCB Comm J-459. 10.12688/f1000research.14509.2 - DOI - PMC - PubMed
-
- Buchta C., Aberle S. W., Allerberger F., Benka B., Gorzer I., Griesmacher A., et al. (2023b). Performance of SARS-CoV-2 nucleic acid amplification testing in Austria as measured by external quality assessment schemes during 3 years of the COVID-19 pandemic: an observational retrospective study. Lancet Microbe 4, e1015–e1023. 10.1016/S2666-5247(23)00286-0 - DOI - PubMed
-
- Buchta C., Camp J. V., Jovanovic J., Puchhammer-Stockl E., Strassl R., Muller M. M., et al. (2022b). Results of a SARS-CoV-2 virus genome detection external quality assessment round focusing on sensitivity of assays and pooling of samples. Clin. Chem. Lab. Med. 60 (8), 1308–1312. 10.1515/cclm-2022-0263 - DOI - PubMed
-
- Buchta C., Camp J. V., Jovanovic J., Radler U., Benka B., Puchhammer-Stockl E., et al. (2022a). Inadequate design of mutation detection panels prevents interpretation of variants of concern: results of an external quality assessment for SARS-CoV-2 variant detection. Clin. Chem. Lab. Med. 60 (2), 291–298. 10.1515/cclm-2021-0889 - DOI - PubMed
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous