

METTL17 coordinates ferroptosis and tumorigenesis by regulating mitochondrial translation in colorectal cancer
- PMID: 38377789
- PMCID: PMC10884776
- DOI: 10.1016/j.redox.2024.103087
METTL17 coordinates ferroptosis and tumorigenesis by regulating mitochondrial translation in colorectal cancer
Abstract
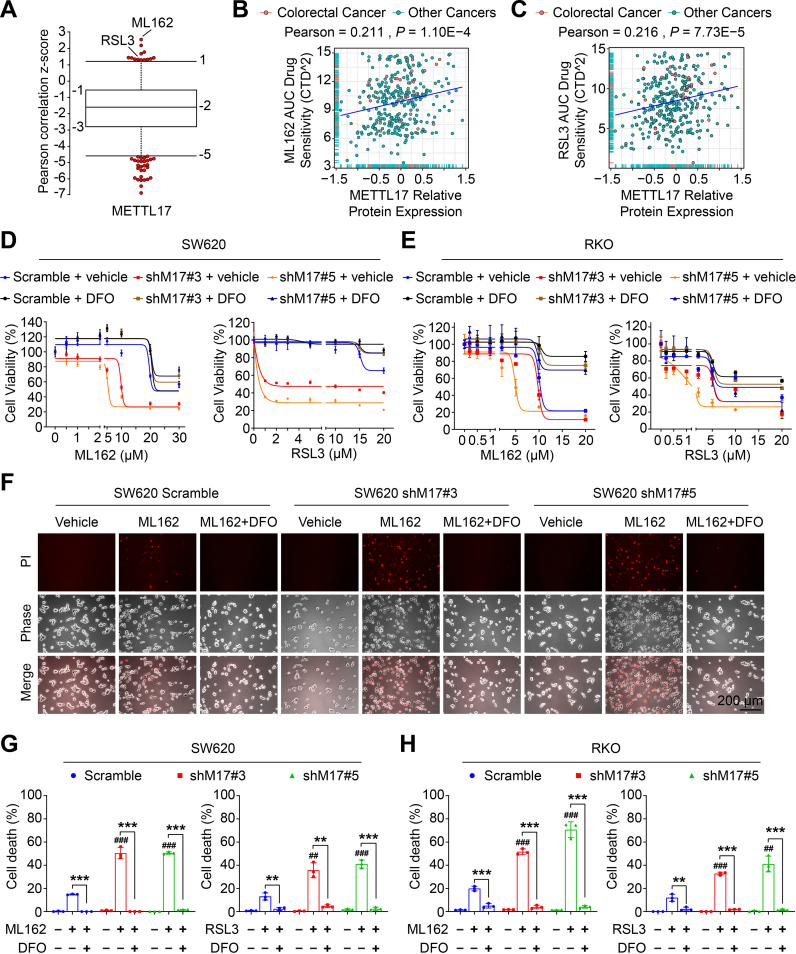
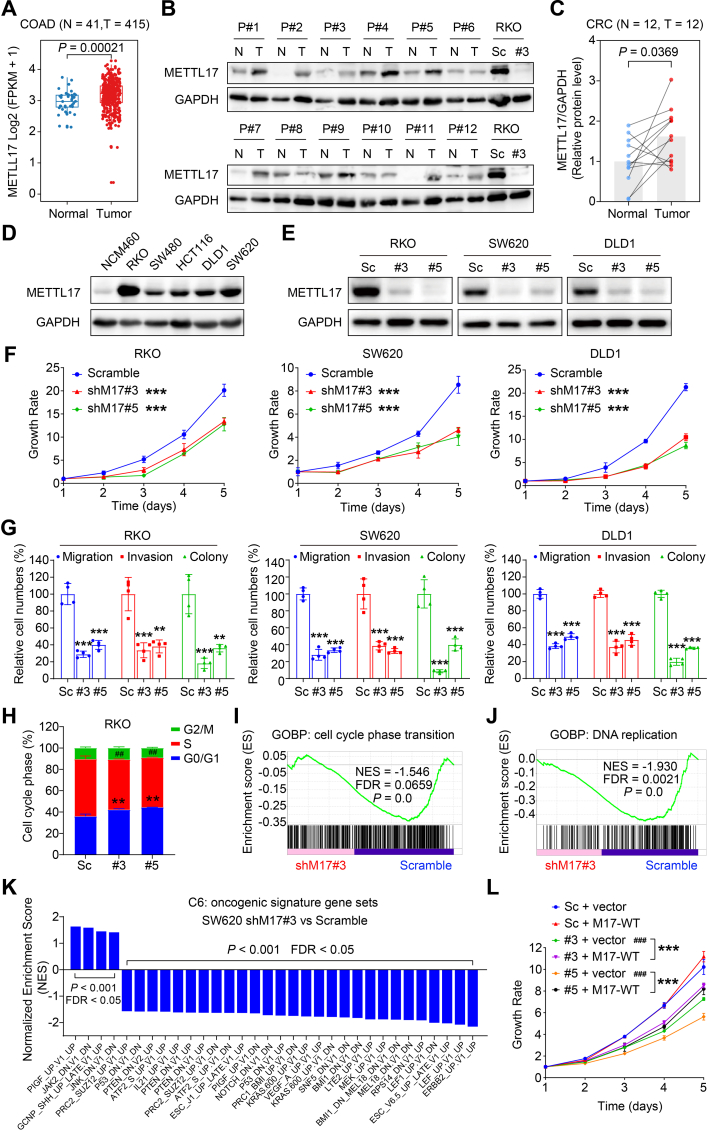
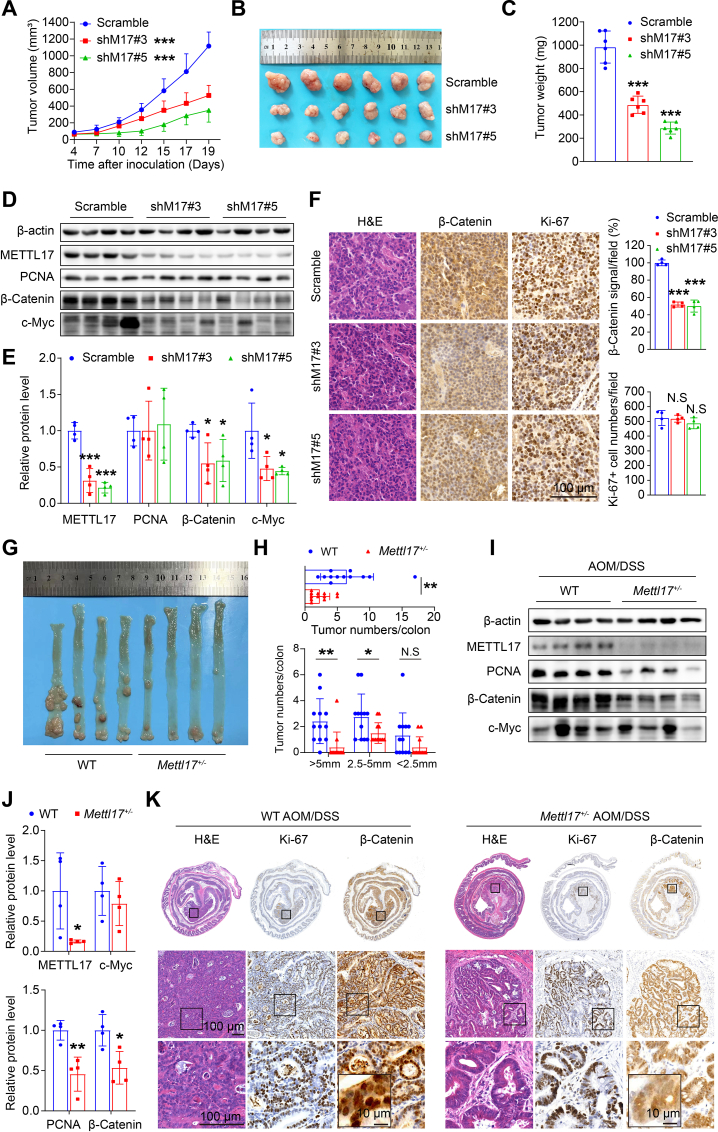
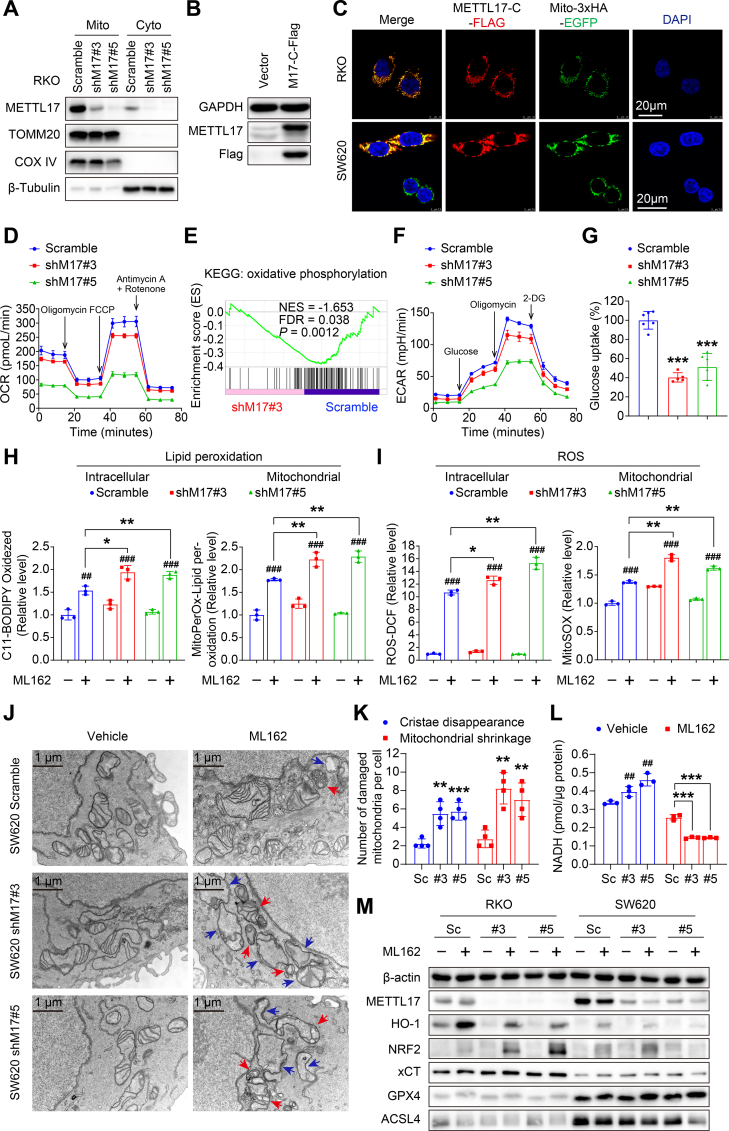
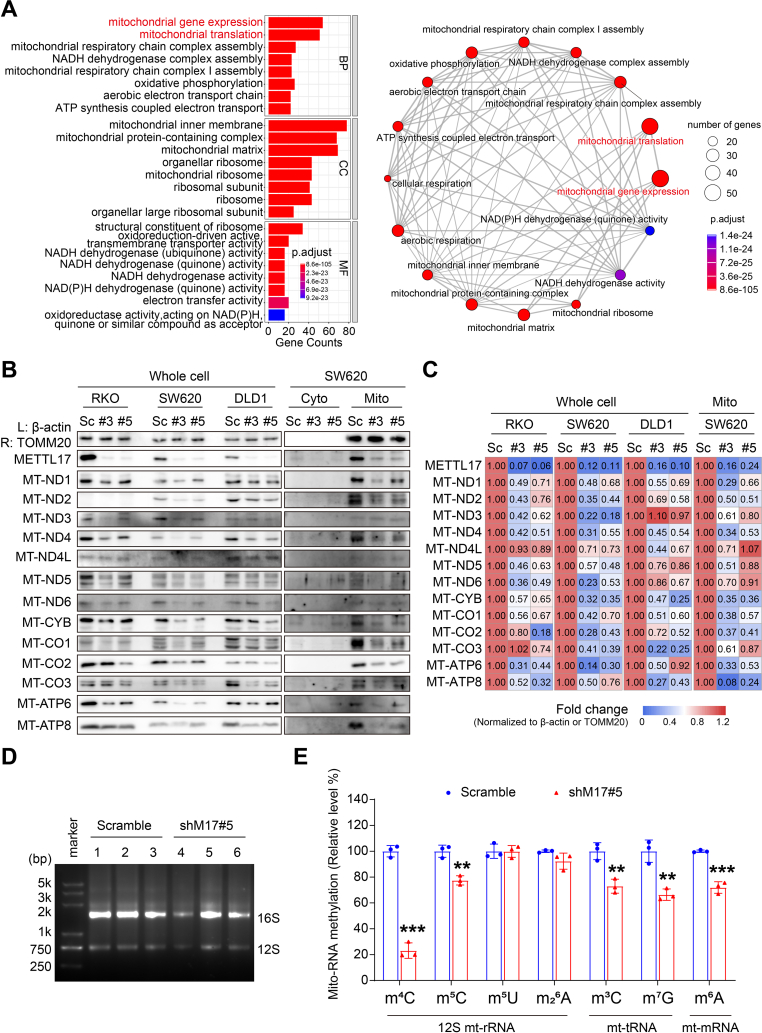
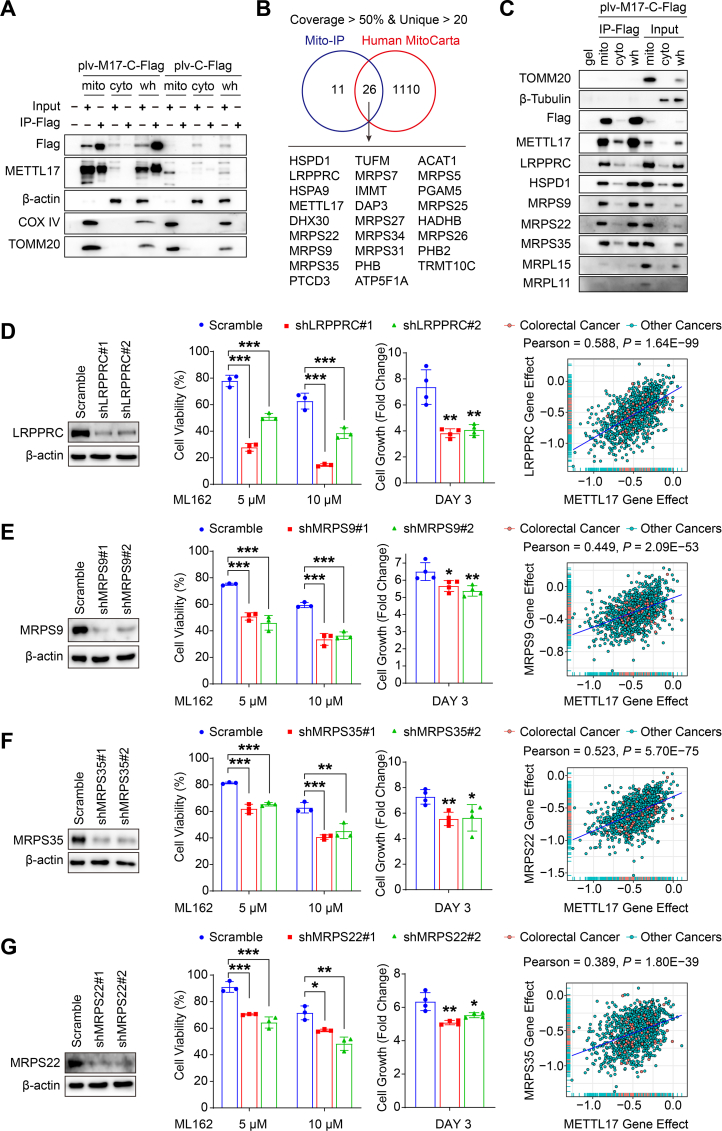
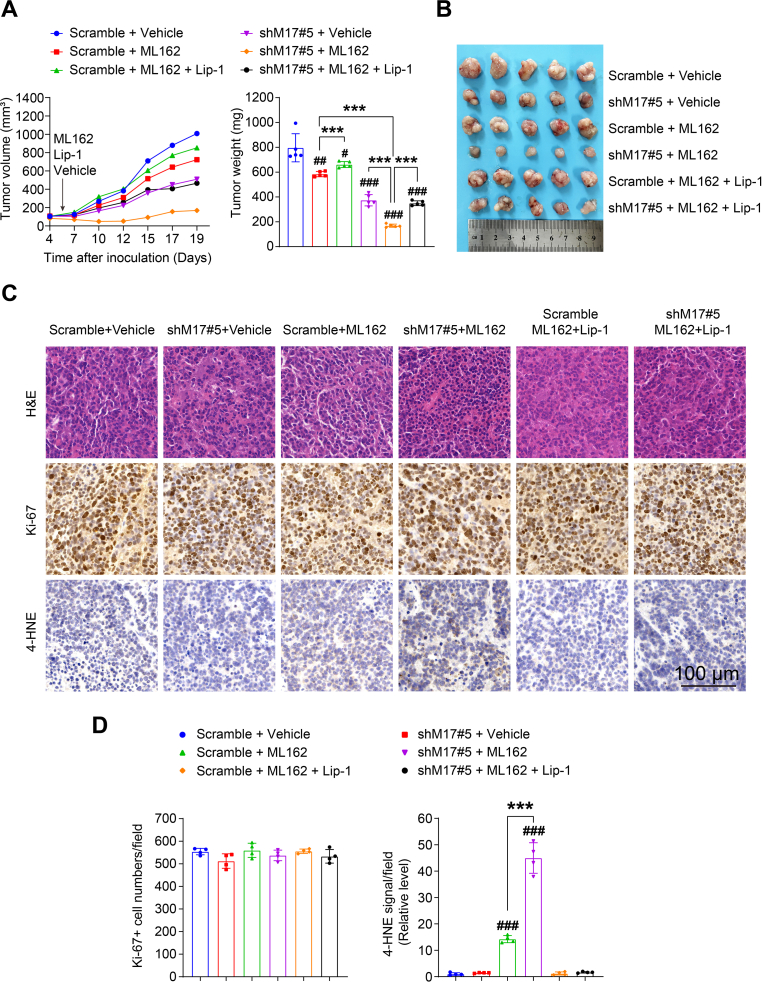
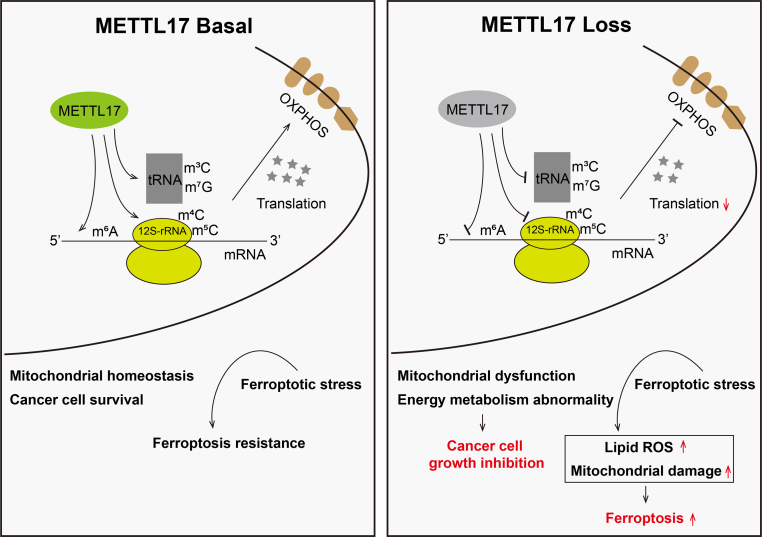
Ferroptosis, an iron-dependent lipid peroxidation-induced form of regulated cell death, shows great promise as a cancer therapy strategy. Despite the critical role of mitochondria in ferroptosis regulation, the underlying mechanisms remain elusive. This study reveals that the mitochondrial protein METTL17 governs mitochondrial function in colorectal cancer (CRC) cells through epigenetic modulation. Bioinformatic analysis establishes that METTL17 expression positively correlates with ferroptosis resistance in cancer cells and is up-regulated in CRC. Depletion of METTL17 sensitizes CRC cells to ferroptosis, impairs cell proliferation, migration, invasion, xenograft tumor growth, and AOM/DSS-induced CRC tumorigenesis. Furthermore, suppression of METTL17 disrupts mitochondrial function, energy metabolism, and enhances intracellular and mitochondrial lipid peroxidation and ROS levels during ferroptotic stress. Mechanistically, METTL17 inhibition significantly reduces mitochondrial RNA methylation, including m4C, m5C, m3C, m7G, and m6A, leading to impaired translation of mitochondrial protein-coding genes. Additionally, the interacting proteins associated with METTL17 are essential for mitochondrial gene expression, and their knockdown sensitizes CRC cells to ferroptosis and inhibits cell proliferation. Notably, combined targeting of METTL17 and ferroptosis in a therapeutic approach effectively suppresses CRC xenograft growth in vivo. This study uncovers the METTL17-mediated defense mechanism for cell survival and ferroptosis in mitochondria, highlighting METTL17 as a potential therapeutic target for CRC.
Keywords: Colorectal cancer (CRC); Ferroptosis; METTL17; Mitochondrial RNA methylation.
Copyright © 2024 The Authors. Published by Elsevier B.V. All rights reserved.
Conflict of interest statement
Declaration of competing interest The authors declare no competing interests.
Figures
Similar articles
-
ATF3-CBS signaling axis coordinates ferroptosis and tumorigenesis in colorectal cancer.Redox Biol. 2024 May;71:103118. doi: 10.1016/j.redox.2024.103118. Epub 2024 Mar 8. Redox Biol. 2024. PMID: 38490069 Free PMC article.
-
SNORA56-mediated pseudouridylation of 28 S rRNA inhibits ferroptosis and promotes colorectal cancer proliferation by enhancing GCLC translation.J Exp Clin Cancer Res. 2023 Dec 5;42(1):331. doi: 10.1186/s13046-023-02906-8. J Exp Clin Cancer Res. 2023. PMID: 38049865 Free PMC article.
-
Musashi-2 Deficiency Triggers Colorectal Cancer Ferroptosis by Downregulating the MAPK Signaling Cascade to Inhibit HSPB1 Phosphorylation.Biol Proced Online. 2023 Dec 1;25(1):32. doi: 10.1186/s12575-023-00222-1. Biol Proced Online. 2023. PMID: 38041016 Free PMC article.
-
Targeting critical pathways in ferroptosis and enhancing antitumor therapy of Platinum drugs for colorectal cancer.Sci Prog. 2023 Jan-Mar;106(1):368504221147173. doi: 10.1177/00368504221147173. Sci Prog. 2023. PMID: 36718538 Free PMC article. Review.
-
Ferroptosis: the balance between death and survival in colorectal cancer.Int J Biol Sci. 2024 Jul 2;20(10):3773-3783. doi: 10.7150/ijbs.96828. eCollection 2024. Int J Biol Sci. 2024. PMID: 39113707 Free PMC article. Review.
Cited by
-
Targeting treatment resistance: unveiling the potential of RNA methylation regulators and TG-101,209 in pan-cancer neoadjuvant therapy.J Exp Clin Cancer Res. 2024 Aug 19;43(1):232. doi: 10.1186/s13046-024-03111-x. J Exp Clin Cancer Res. 2024. PMID: 39160604 Free PMC article.
-
Ferroptosis in cancer: revealing the multifaceted functions of mitochondria.Cell Mol Life Sci. 2025 Jul 17;82(1):277. doi: 10.1007/s00018-025-05812-8. Cell Mol Life Sci. 2025. PMID: 40676247 Free PMC article. Review.
-
Dihydroartemisinin Sensitizes Lung Cancer Cells to Cisplatin Treatment by Upregulating ZIP14 Expression and Inducing Ferroptosis.Cancer Med. 2024 Oct;13(19):e70271. doi: 10.1002/cam4.70271. Cancer Med. 2024. PMID: 39394878 Free PMC article.
-
The cytochrome oxidase defect in ISC-depleted yeast is caused by impaired iron-sulfur cluster maturation of the mitoribosome assembly factor Rsm22.FEBS Lett. 2025 Aug;599(16):2301-2317. doi: 10.1002/1873-3468.70129. Epub 2025 Aug 6. FEBS Lett. 2025. PMID: 40768618 Free PMC article.
-
N7-methylguanosine modification in cancers: from mechanisms to therapeutic potential.J Hematol Oncol. 2025 Jan 29;18(1):12. doi: 10.1186/s13045-025-01665-7. J Hematol Oncol. 2025. PMID: 39881381 Free PMC article. Review.
References
-
- Chen X., Kang R., Kroemer G., Tang D.L. Broadening horizons: the role of ferroptosis in cancer. Nat. Rev. Clin. Oncol. 2021;18:280–296. - PubMed
-
- Hassannia B., Vandenabeele P., Vanden Berghe T. Targeting ferroptosis to iron out cancer. Cancer Cell. 2019;35:830–849. - PubMed
-
- Liang C., Zhang X.L., Yang M.S., Dong X.C. Recent progress in ferroptosis inducers for cancer therapy. Adv. Mater. 2019;31 - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
