

Endoplasmic reticular stress as an emerging therapeutic target for chronic pain: a narrative review
- PMID: 38378384
- PMCID: PMC10925894
- DOI: 10.1016/j.bja.2024.01.007
Endoplasmic reticular stress as an emerging therapeutic target for chronic pain: a narrative review
Abstract
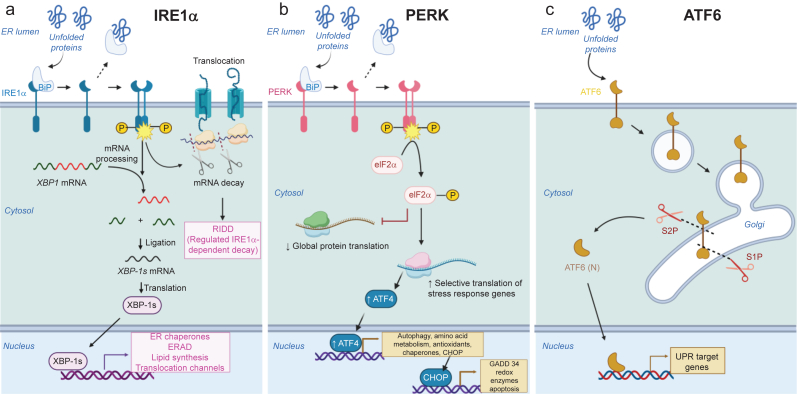
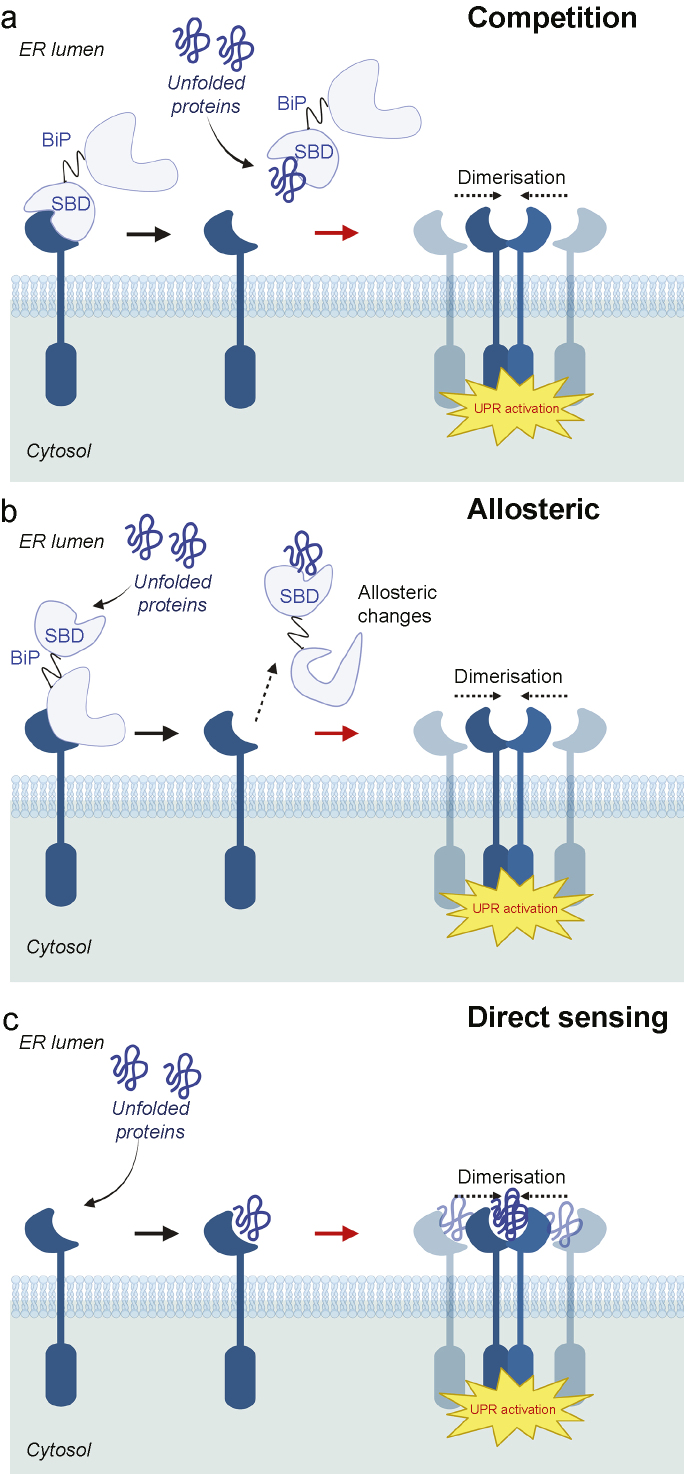
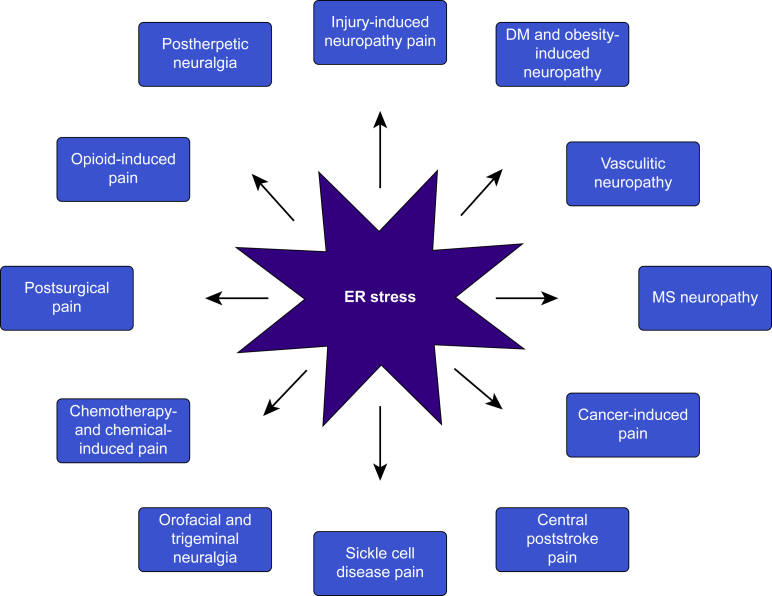
Chronic pain is a severely debilitating condition with enormous socioeconomic costs. Current treatment regimens with nonsteroidal anti-inflammatory drugs (NSAIDs), steroids, or opioids have been largely unsatisfactory with uncertain benefits or severe long-term side effects. This is mainly because chronic pain has a multifactorial aetiology. Although conventional pain medications can alleviate pain by keeping several dysfunctional pathways under control, they can mask other underlying pathological causes, ultimately worsening nerve pathologies and pain outcome. Recent preclinical studies have shown that endoplasmic reticulum (ER) stress could be a central hub for triggering multiple molecular cascades involved in the development of chronic pain. Several ER stress inhibitors and unfolded protein response modulators, which have been tested in randomised clinical trials or apprpoved by the US Food and Drug Administration for other chronic diseases, significantly alleviated hyperalgesia in multiple preclinical pain models. Although the role of ER stress in neurodegenerative disorders, metabolic disorders, and cancer has been well established, research on ER stress and chronic pain is still in its infancy. Here, we critically analyse preclinical studies and explore how ER stress can mechanistically act as a central node to drive development and progression of chronic pain. We also discuss therapeutic prospects, benefits, and pitfalls of using ER stress inhibitors and unfolded protein response modulators for managing intractable chronic pain. In the future, targeting ER stress to impact multiple molecular networks might be an attractive therapeutic strategy against chronic pain refractory to steroids, NSAIDs, or opioids. This novel therapeutic strategy could provide solutions for the opioid crisis and public health challenge.
Keywords: animal model; apoptosis; chronic pain; endoplasmic reticulum stress; inflammation; ion channel; mitochondrial dysfunction; unfolded protein response.
Copyright © 2024 British Journal of Anaesthesia. Published by Elsevier Ltd. All rights reserved.
Conflict of interest statement
The authors declare that they have no conflicts of interest.
Figures
Similar articles
-
Unraveling the Connection: Pain and Endoplasmic Reticulum Stress.Int J Mol Sci. 2024 May 3;25(9):4995. doi: 10.3390/ijms25094995. Int J Mol Sci. 2024. PMID: 38732214 Free PMC article. Review.
-
Role of ER Stress Mediated Unfolded Protein Responses and ER Stress Inhibitors in the Pathogenesis of Inflammatory Bowel Disease.Dig Dis Sci. 2022 Dec;67(12):5392-5406. doi: 10.1007/s10620-022-07467-y. Epub 2022 Mar 22. Dig Dis Sci. 2022. PMID: 35318552 Review.
-
Endoplasmic reticulum stress and unfolded protein response in diaphragm muscle dysfunction of patients with stable chronic obstructive pulmonary disease.J Appl Physiol (1985). 2019 Jun 1;126(6):1572-1586. doi: 10.1152/japplphysiol.00670.2018. Epub 2019 Apr 18. J Appl Physiol (1985). 2019. PMID: 30998124
-
Endoplasmic reticulum stress and therapeutic strategies in metabolic, neurodegenerative diseases and cancer.Mol Med. 2024 Mar 20;30(1):40. doi: 10.1186/s10020-024-00808-9. Mol Med. 2024. PMID: 38509524 Free PMC article. Review.
-
Oxicam-derived non-steroidal anti-inflammatory drugs suppress 1-methyl-4-phenyl pyridinium-induced cell death via repression of endoplasmic reticulum stress response and mitochondrial dysfunction in SH-SY5Y cells.Biochem Biophys Res Commun. 2018 Sep 18;503(4):2963-2969. doi: 10.1016/j.bbrc.2018.08.078. Epub 2018 Aug 11. Biochem Biophys Res Commun. 2018. PMID: 30107908
Cited by
-
Inhibition of Soluble Epoxide Hydrolase Prevents Docetaxel-Induced Painful Peripheral Neuropathy.Int J Mol Sci. 2025 Jun 12;26(12):5630. doi: 10.3390/ijms26125630. Int J Mol Sci. 2025. PMID: 40565091 Free PMC article.
-
Unraveling the AMPK-SIRT1-FOXO Pathway: The In-Depth Analysis and Breakthrough Prospects of Oxidative Stress-Induced Diseases.Antioxidants (Basel). 2025 Jan 9;14(1):70. doi: 10.3390/antiox14010070. Antioxidants (Basel). 2025. PMID: 39857404 Free PMC article. Review.
-
Nitroxidative Stress, Cell-Signaling Pathways, and Manganese Porphyrins: Therapeutic Potential in Neuropathic Pain.Int J Mol Sci. 2025 Feb 26;26(5):2050. doi: 10.3390/ijms26052050. Int J Mol Sci. 2025. PMID: 40076672 Free PMC article. Review.
-
Unraveling the Connection: Pain and Endoplasmic Reticulum Stress.Int J Mol Sci. 2024 May 3;25(9):4995. doi: 10.3390/ijms25094995. Int J Mol Sci. 2024. PMID: 38732214 Free PMC article. Review.
-
Molecular Mechanisms of Chronic Pain and Therapeutic Interventions.MedComm (2020). 2025 Aug 7;6(8):e70325. doi: 10.1002/mco2.70325. eCollection 2025 Aug. MedComm (2020). 2025. PMID: 40787071 Free PMC article. Review.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
