

Resistance of HNSCC cell models to pan-FGFR inhibition depends on the EMT phenotype associating with clinical outcome
- PMID: 38378518
- PMCID: PMC10880239
- DOI: 10.1186/s12943-024-01954-8
Resistance of HNSCC cell models to pan-FGFR inhibition depends on the EMT phenotype associating with clinical outcome
Abstract
Background: Focal adhesion signaling involving receptor tyrosine kinases (RTK) and integrins co-controls cancer cell survival and therapy resistance. However, co-dependencies between these receptors and therapeutically exploitable vulnerabilities remain largely elusive in HPV-negative head and neck squamous cell carcinoma (HNSCC).
Methods: The cytotoxic and radiochemosensitizing potential of targeting 10 RTK and β1 integrin was determined in up to 20 3D matrix-grown HNSCC cell models followed by drug screening and patient-derived organoid validation. RNA sequencing and protein-based biochemical assays were performed for molecular characterization. Bioinformatically identified transcriptomic signatures were applied to patient cohorts.
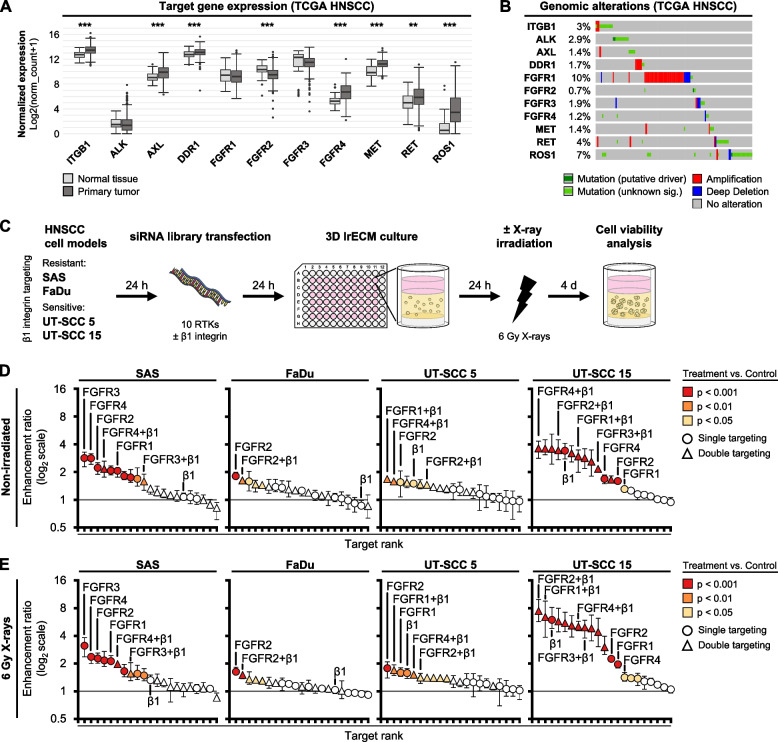
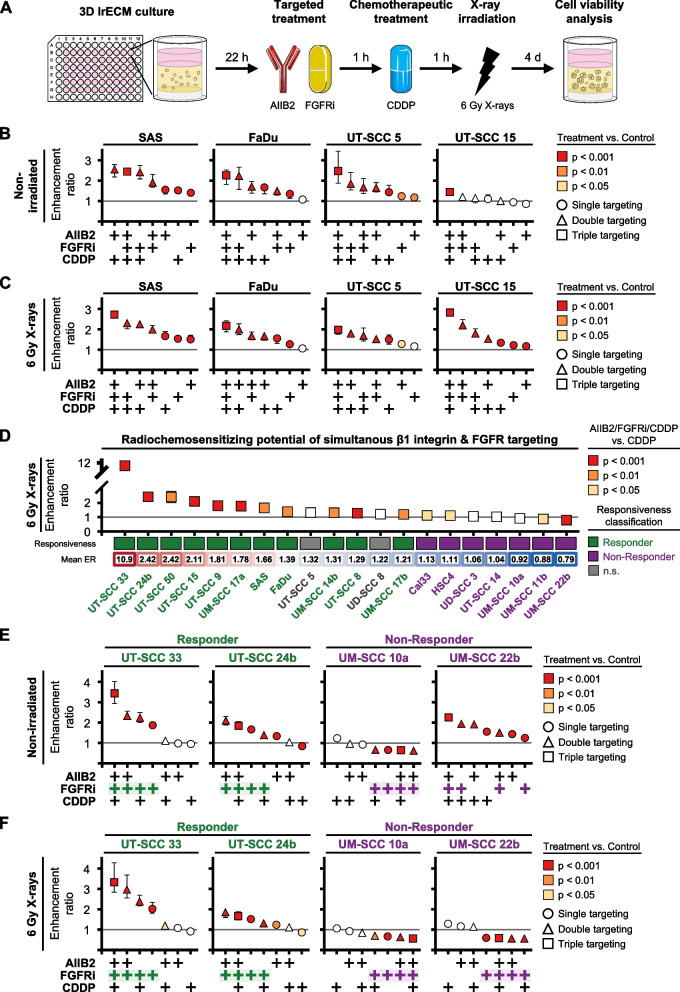
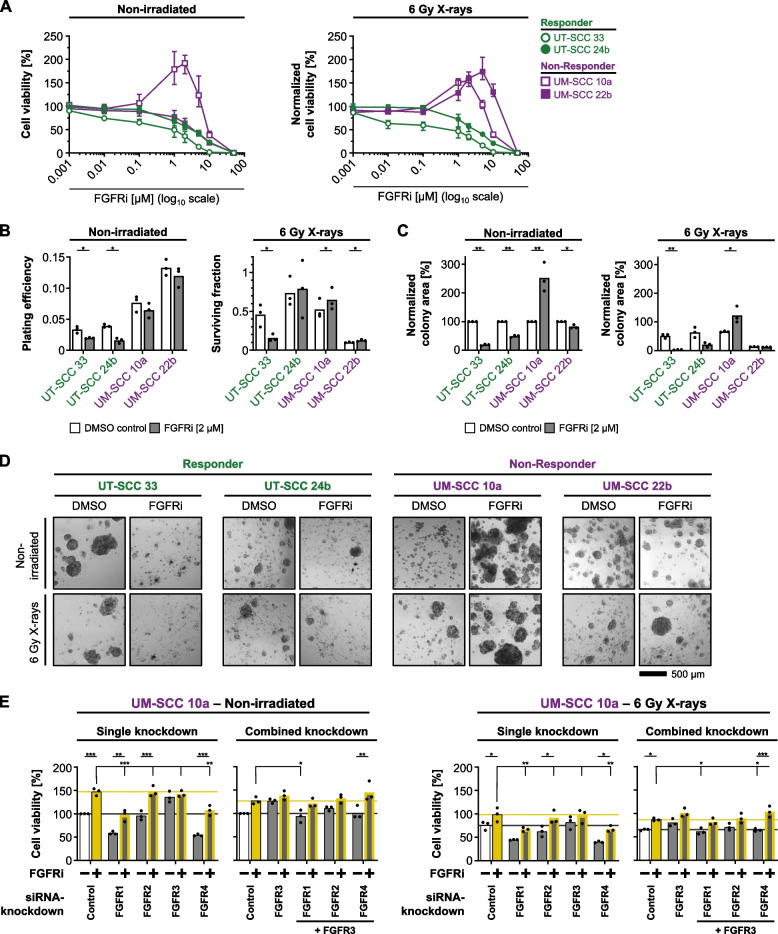
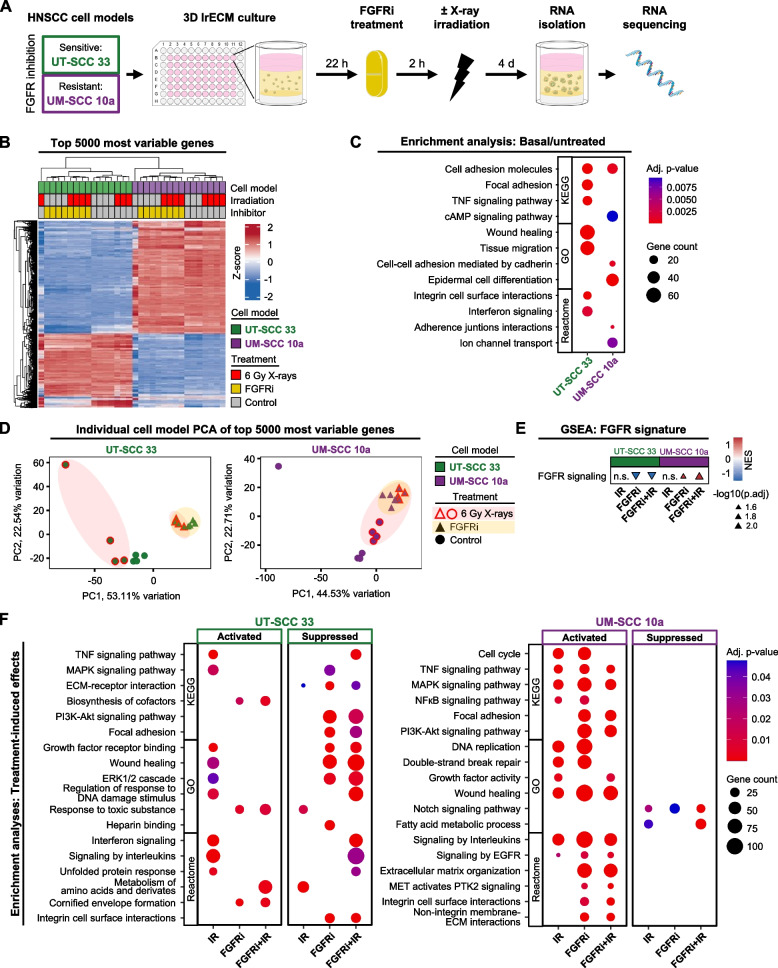
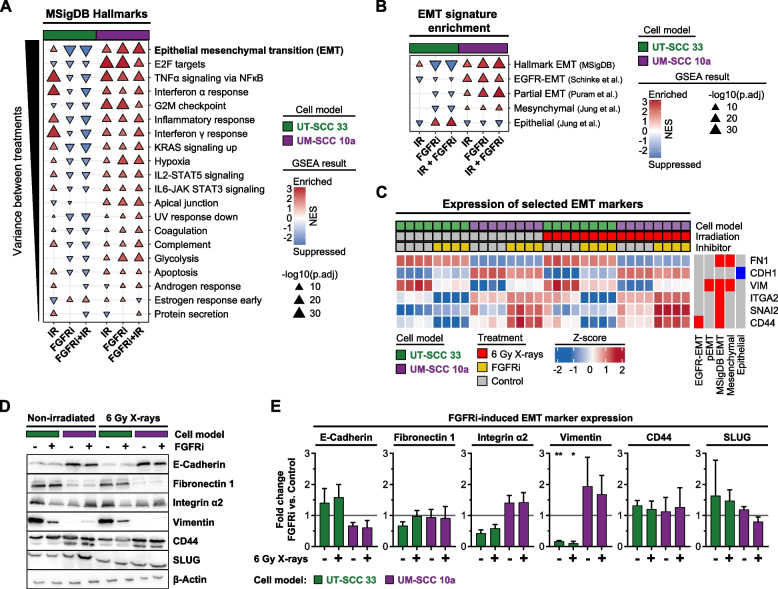
Results: Fibroblast growth factor receptor (FGFR 1-4) targeting exhibited the strongest cytotoxic and radiosensitizing effects as monotherapy and combined with β1 integrin inhibition, exceeding the efficacy of the other RTK studied. Pharmacological pan-FGFR inhibition elicited responses ranging from cytotoxicity/radiochemosensitization to resistance/radiation protection. RNA sequence analysis revealed a mesenchymal-to-epithelial transition (MET) in sensitive cell models, whereas resistant cell models exhibited a partial epithelial-to-mesenchymal transition (EMT). Accordingly, inhibition of EMT-associated kinases such as EGFR caused reduced adaptive resistance and enhanced (radio)sensitization to FGFR inhibition cell model- and organoid-dependently. Transferring the EMT-associated transcriptomic profiles to HNSCC patient cohorts not only demonstrated their prognostic value but also provided a conclusive validation of the presence of EGFR-related vulnerabilities that can be strategically exploited for therapeutic interventions.
Conclusions: This study demonstrates that pan-FGFR inhibition elicits a beneficial radiochemosensitizing and a detrimental radioprotective potential in HNSCC cell models. Adaptive EMT-associated resistance appears to be of clinical importance, and we provide effective molecular approaches to exploit this therapeutically.
Keywords: Adaptive resistance; Epidermal growth factor receptor; Epithelial-to-mesenchymal transition; Fibroblast growth factor receptor; HNSCC; Radioprotection; Radiosensitization; β1 integrin.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures



References
-
- Johnson DE, Burtness B, Leemans CR, Lui VWY, Bauman JE, Grandis JR. Head and neck squamous cell carcinoma. Nat Rev Dis Prim 2020 61. 2020 [cited 2023 Aug 1];6:1–22. Available from: https://www.nature.com/articles/s41572-020-00224-3 - PMC - PubMed
-
- Cramer JD, Burtness B, Le QT, Ferris RL. The changing therapeutic landscape of head and neck cancer. Nat Rev Clin Oncol 2019 1611. 2019 [cited 2023 Aug 2];16:669–83. Available from: https://www.nature.com/articles/s41571-019-0227-z - PubMed
-
- Vasan N, Baselga J, Hyman DM. A view on drug resistance in cancer. Nat 2019 5757782. 2019 [cited 2022 Dec 8];575:299–309. Available from: https://www.nature.com/articles/s41586-019-1730-1 - PMC - PubMed
-
- Huang C, Chen L, Savage SR, Eguez RV, Dou Y, Li Y, et al. Proteogenomic insights into the biology and treatment of HPV-negative head and neck squamous cell carcinoma. Cancer Cell. 2021 [cited 2023 Aug 2];39:361–379.e16. Available from: http://www.cell.com/article/S1535610820306553/fulltext - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
