

Multimodal and spatially resolved profiling identifies distinct patterns of T cell infiltration in nodal B cell lymphoma entities
- PMID: 38379051
- PMCID: PMC10940160
- DOI: 10.1038/s41556-024-01358-2
Multimodal and spatially resolved profiling identifies distinct patterns of T cell infiltration in nodal B cell lymphoma entities
Abstract
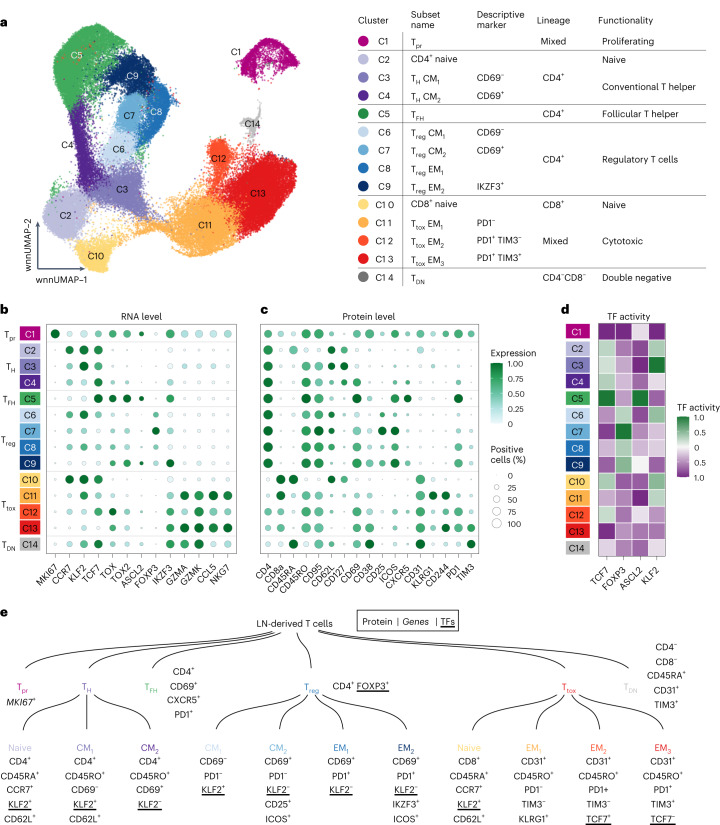
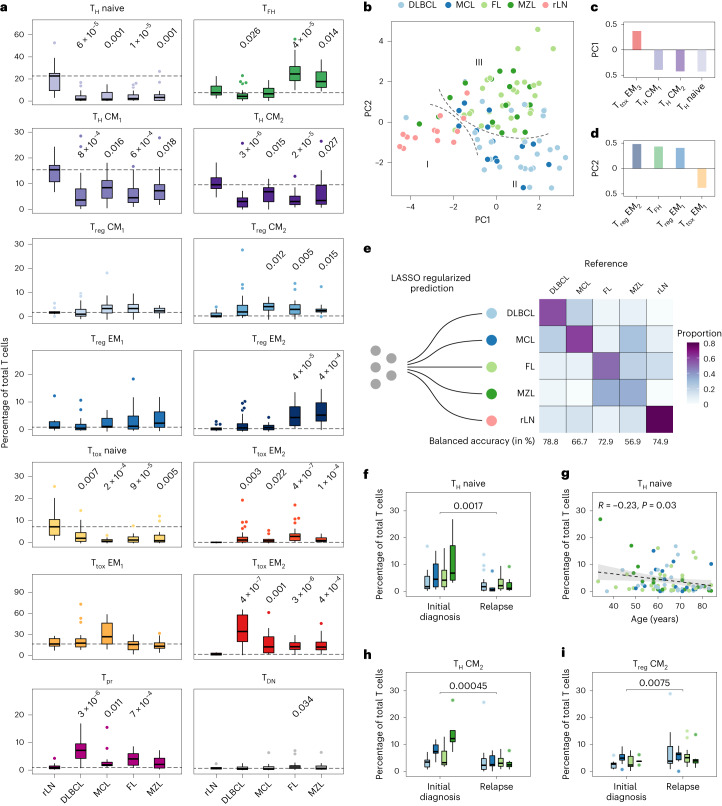
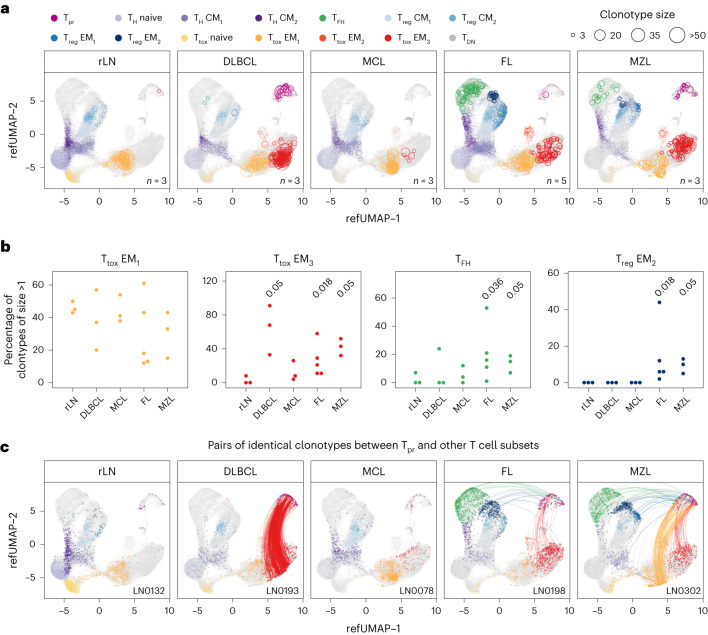
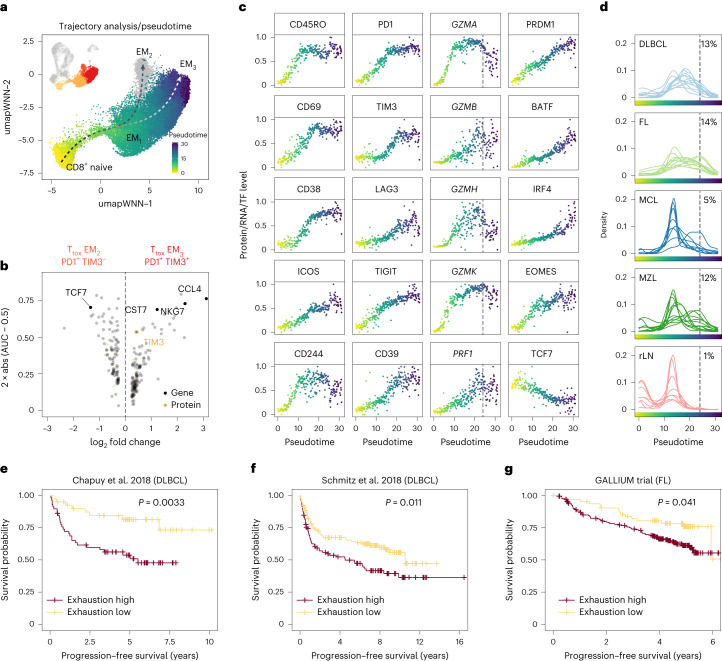
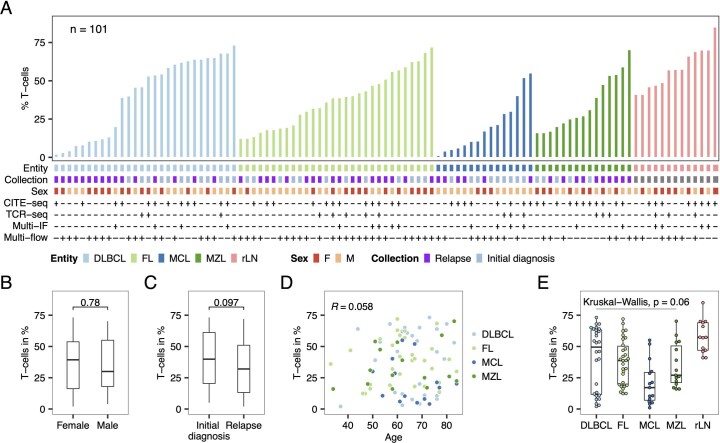
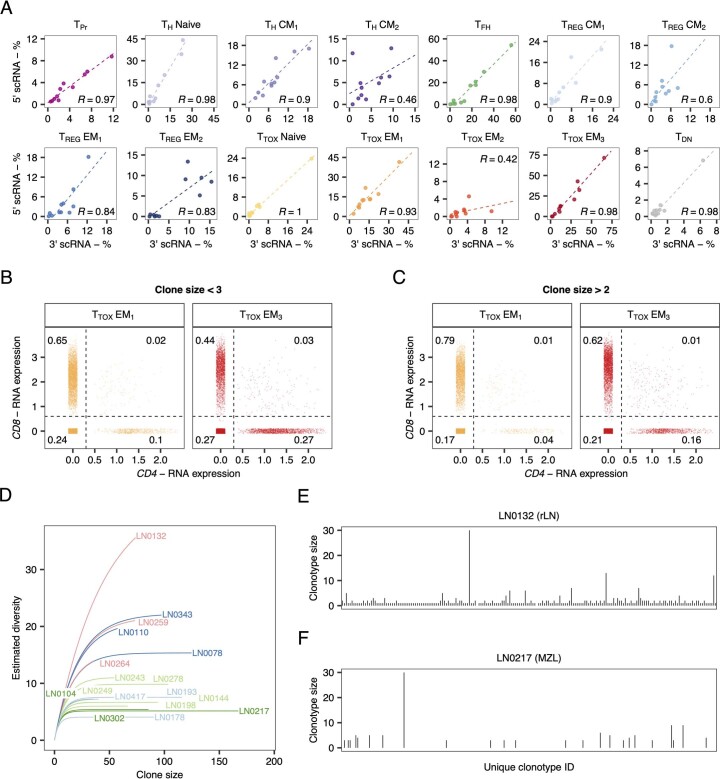
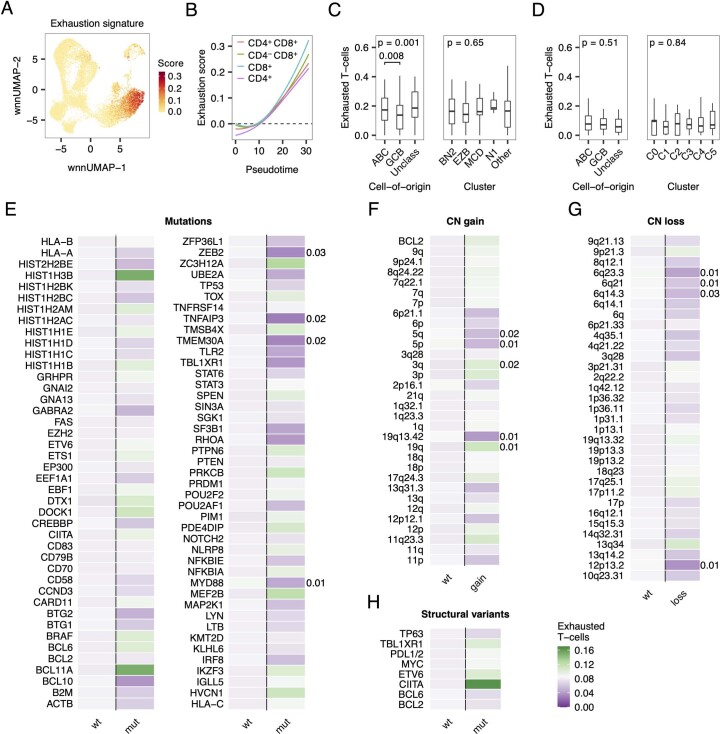
The redirection of T cells has emerged as an attractive therapeutic principle in B cell non-Hodgkin lymphoma (B-NHL). However, a detailed characterization of lymphoma-infiltrating T cells across B-NHL entities is missing. Here we present an in-depth T cell reference map of nodal B-NHL, based on cellular indexing of transcriptomes and epitopes, T cell receptor sequencing, flow cytometry and multiplexed immunofluorescence applied to 101 lymph nodes from patients with diffuse large B cell, mantle cell, follicular or marginal zone lymphoma, and from healthy controls. This multimodal resource revealed quantitative and spatial aberrations of the T cell microenvironment across and within B-NHL entities. Quantitative differences in PD1+ TCF7- cytotoxic T cells, T follicular helper cells or IKZF3+ regulatory T cells were linked to their clonal expansion. The abundance of PD1+ TCF7- cytotoxic T cells was associated with poor survival. Our study portrays lymphoma-infiltrating T cells with unprecedented comprehensiveness and provides a unique resource for the investigation of lymphoma biology and prognosis.
© 2024. The Author(s).
Conflict of interest statement
C.M.S. is a scientific advisor to, has stock options in and has received research funding from Enable Medicine, Inc. G.P.N. is a co-founder and stockholder of Akoya Biosciences, Inc. and inventor on patent US9909167 (On-slide staining by primer extension). The remaining authors declare no competing interests.
Figures










References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
