

Selective CDK7 Inhibition Suppresses Cell Cycle Progression and MYC Signaling While Enhancing Apoptosis in Therapy-resistant Estrogen Receptor-positive Breast Cancer
- PMID: 38381406
- PMCID: PMC11061603
- DOI: 10.1158/1078-0432.CCR-23-2975
Selective CDK7 Inhibition Suppresses Cell Cycle Progression and MYC Signaling While Enhancing Apoptosis in Therapy-resistant Estrogen Receptor-positive Breast Cancer
Abstract
Purpose: Resistance to endocrine therapy (ET) and CDK4/6 inhibitors (CDK4/6i) is a clinical challenge in estrogen receptor (ER)-positive (ER+) breast cancer. Cyclin-dependent kinase 7 (CDK7) is a candidate target in endocrine-resistant ER+ breast cancer models and selective CDK7 inhibitors (CDK7i) are in clinical development for the treatment of ER+ breast cancer. Nonetheless, the precise mechanisms responsible for the activity of CDK7i in ER+ breast cancer remain elusive. Herein, we sought to unravel these mechanisms.
Experimental design: We conducted multi-omic analyses in ER+ breast cancer models in vitro and in vivo, including models with different genetic backgrounds. We also performed genome-wide CRISPR/Cas9 knockout screens to identify potential therapeutic vulnerabilities in CDK4/6i-resistant models.
Results: We found that the on-target antitumor effects of CDK7 inhibition in ER+ breast cancer are in part p53 dependent, and involve cell cycle inhibition and suppression of c-Myc. Moreover, CDK7 inhibition exhibited cytotoxic effects, distinctive from the cytostatic nature of ET and CDK4/6i. CDK7 inhibition resulted in suppression of ER phosphorylation at S118; however, long-term CDK7 inhibition resulted in increased ER signaling, supporting the combination of ET with a CDK7i. Finally, genome-wide CRISPR/Cas9 knockout screens identified CDK7 and MYC signaling as putative vulnerabilities in CDK4/6i resistance, and CDK7 inhibition effectively inhibited CDK4/6i-resistant models.
Conclusions: Taken together, these findings support the clinical investigation of selective CDK7 inhibition combined with ET to overcome treatment resistance in ER+ breast cancer. In addition, our study highlights the potential of increased c-Myc activity and intact p53 as predictors of sensitivity to CDK7i-based treatments.
©2024 The Authors; Published by the American Association for Cancer Research.
Figures

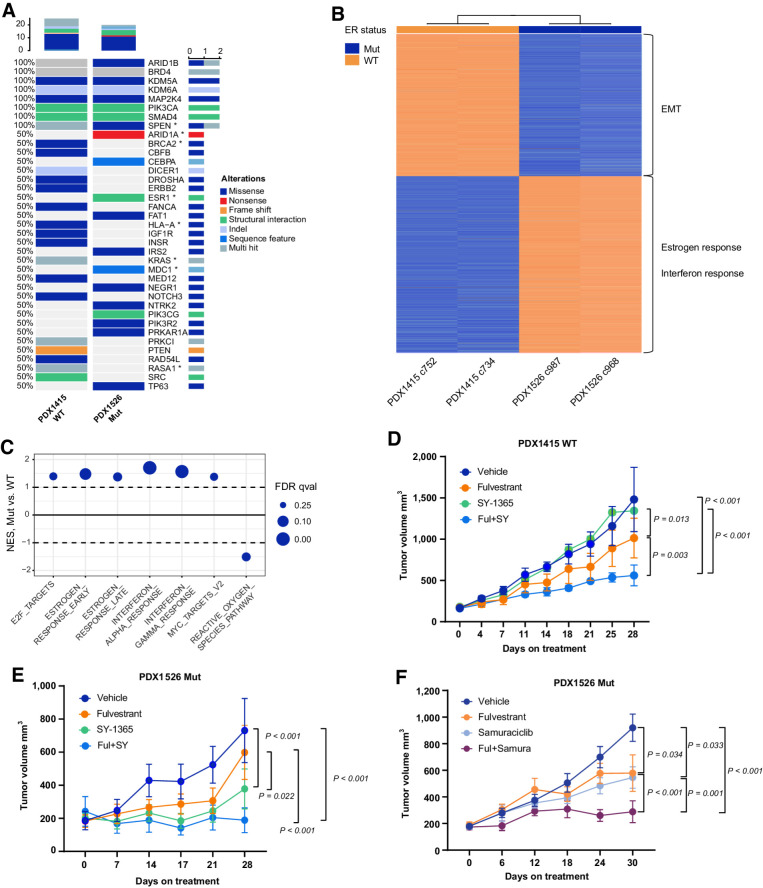




References
-
- Mao P, Cohen O, Kowalski KJ, Kusiel JG, Buendia-Buendia JE, Cuoco MS, et al. . Acquired FGFR and FGF alterations confer resistance to estrogen receptor (ER) targeted therapy in ER(+) metastatic breast cancer. Clin Cancer Res 2020;26:5974–89. - PubMed
-
- Grinshpun A, Sandusky ZM, Jeselsohn R. The clinical utility of ESR1 mutations in hormone receptor-positive, HER2-negative advanced breast cancer. Hematol Oncol Clin North Am 2023;37:169–81. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
