

Prevalence of persistent SARS-CoV-2 in a large community surveillance study
- PMID: 38383783
- PMCID: PMC10901734
- DOI: 10.1038/s41586-024-07029-4
Prevalence of persistent SARS-CoV-2 in a large community surveillance study
Abstract
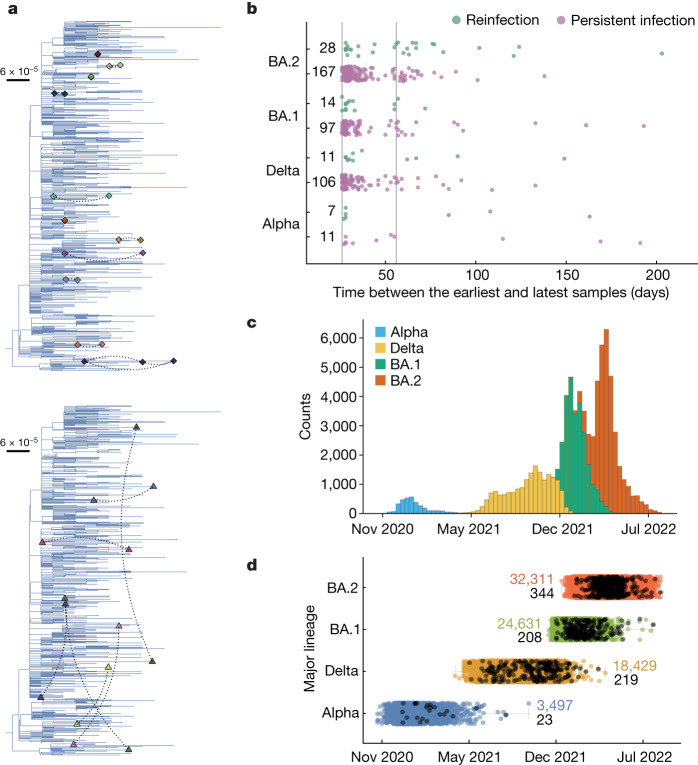
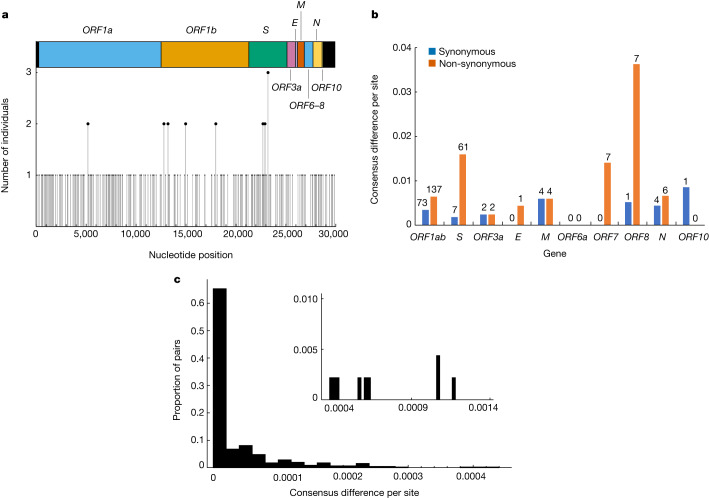
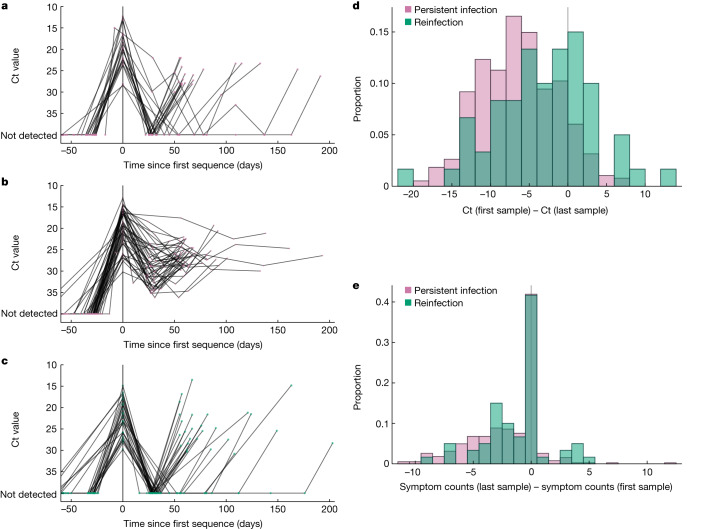
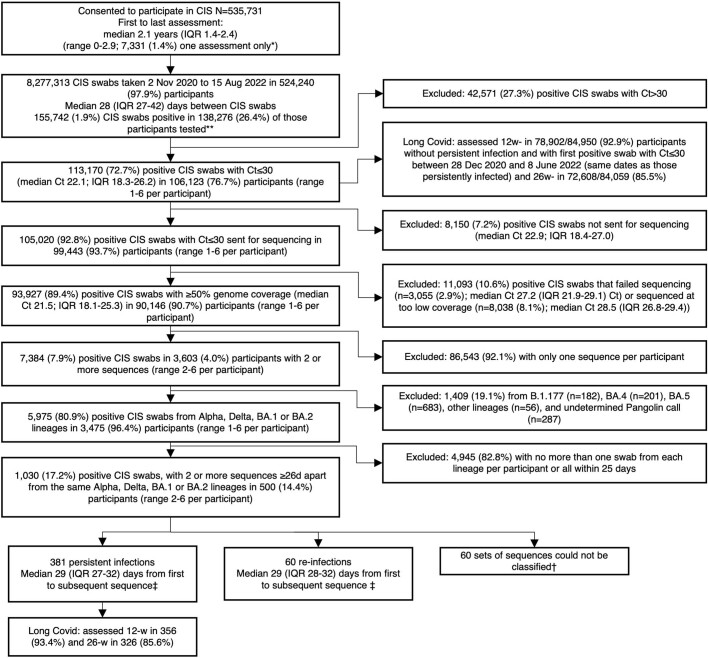
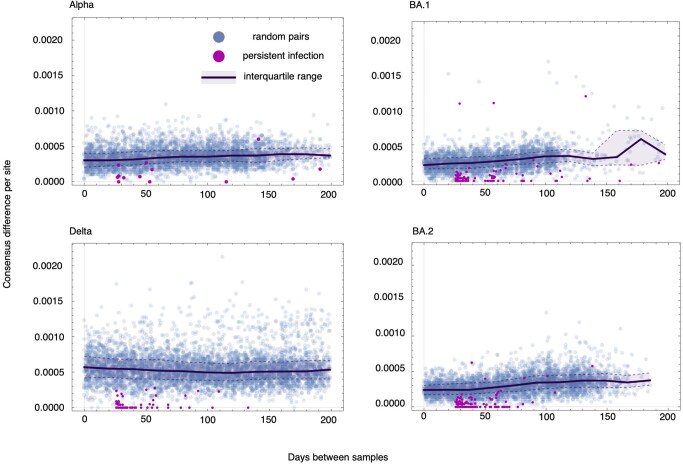
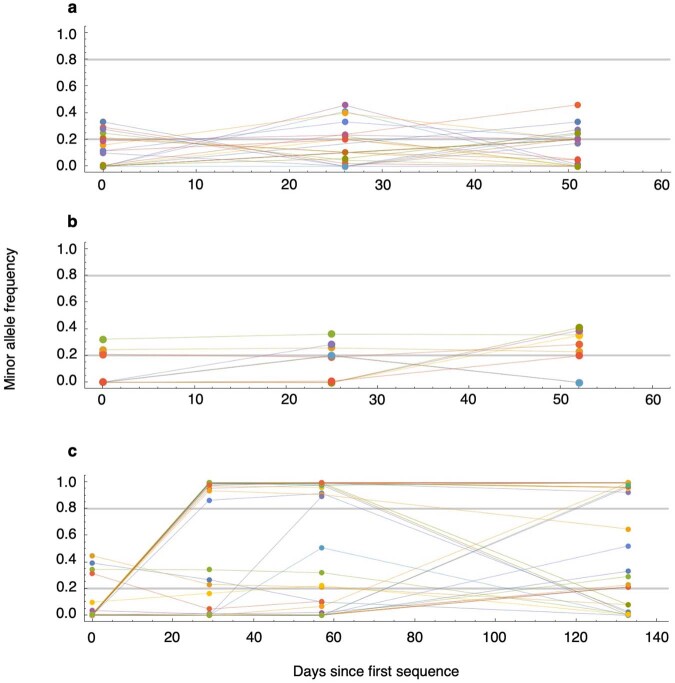
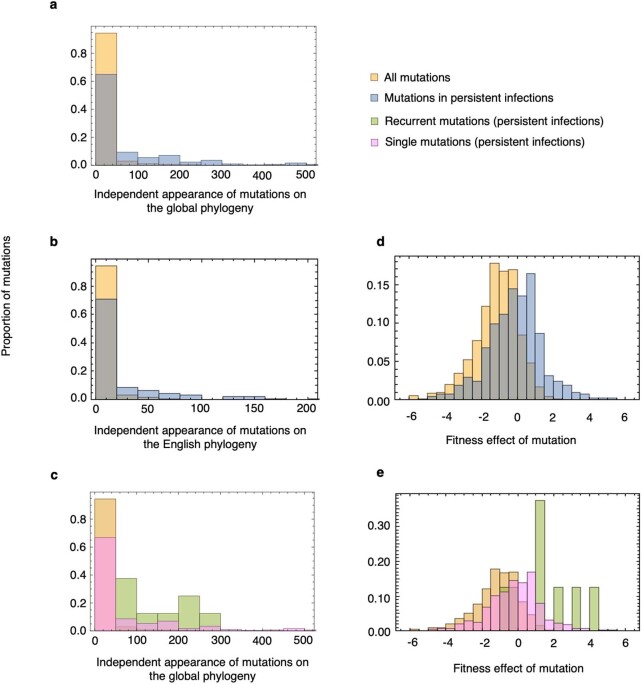
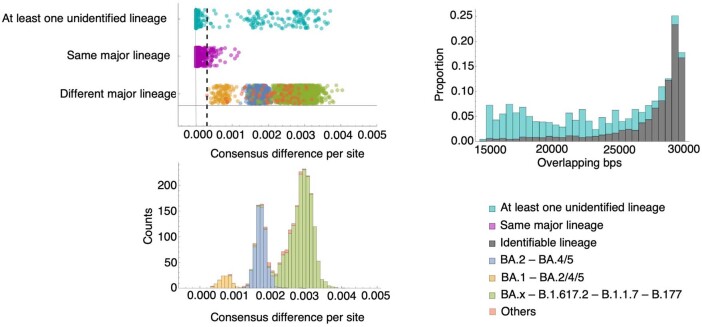
Persistent SARS-CoV-2 infections may act as viral reservoirs that could seed future outbreaks1-5, give rise to highly divergent lineages6-8 and contribute to cases with post-acute COVID-19 sequelae (long COVID)9,10. However, the population prevalence of persistent infections, their viral load kinetics and evolutionary dynamics over the course of infections remain largely unknown. Here, using viral sequence data collected as part of a national infection survey, we identified 381 individuals with SARS-CoV-2 RNA at high titre persisting for at least 30 days, of which 54 had viral RNA persisting at least 60 days. We refer to these as 'persistent infections' as available evidence suggests that they represent ongoing viral replication, although the persistence of non-replicating RNA cannot be ruled out in all. Individuals with persistent infection had more than 50% higher odds of self-reporting long COVID than individuals with non-persistent infection. We estimate that 0.1-0.5% of infections may become persistent with typically rebounding high viral loads and last for at least 60 days. In some individuals, we identified many viral amino acid substitutions, indicating periods of strong positive selection, whereas others had no consensus change in the sequences for prolonged periods, consistent with weak selection. Substitutions included mutations that are lineage defining for SARS-CoV-2 variants, at target sites for monoclonal antibodies and/or are commonly found in immunocompromised people11-14. This work has profound implications for understanding and characterizing SARS-CoV-2 infection, epidemiology and evolution.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
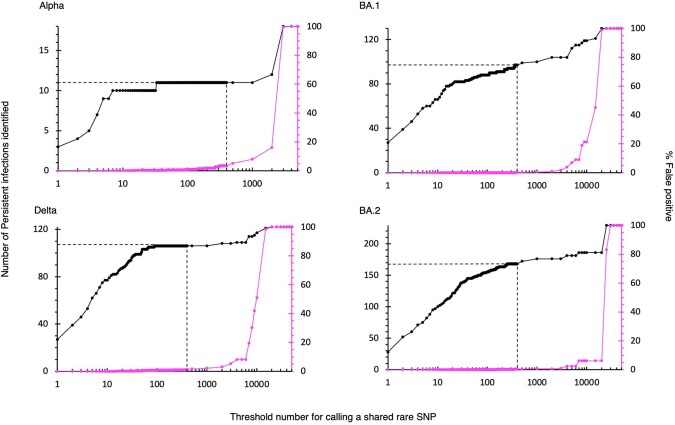
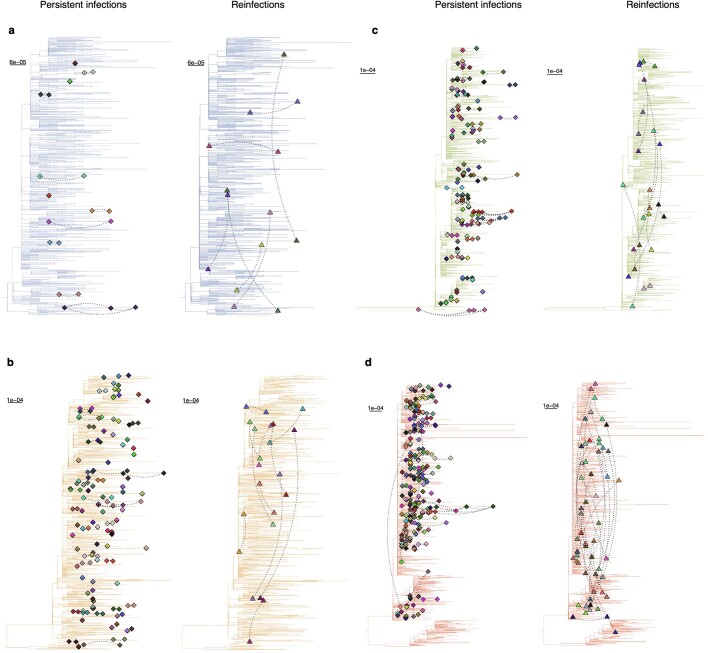
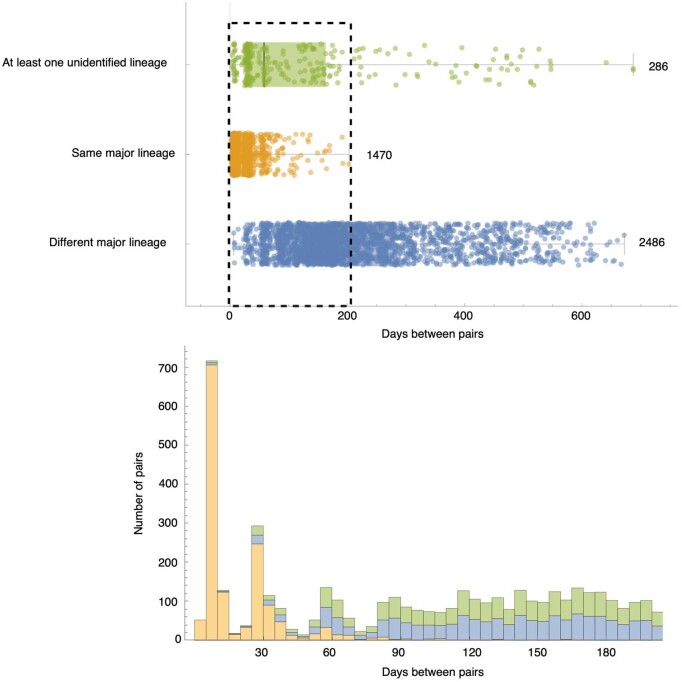
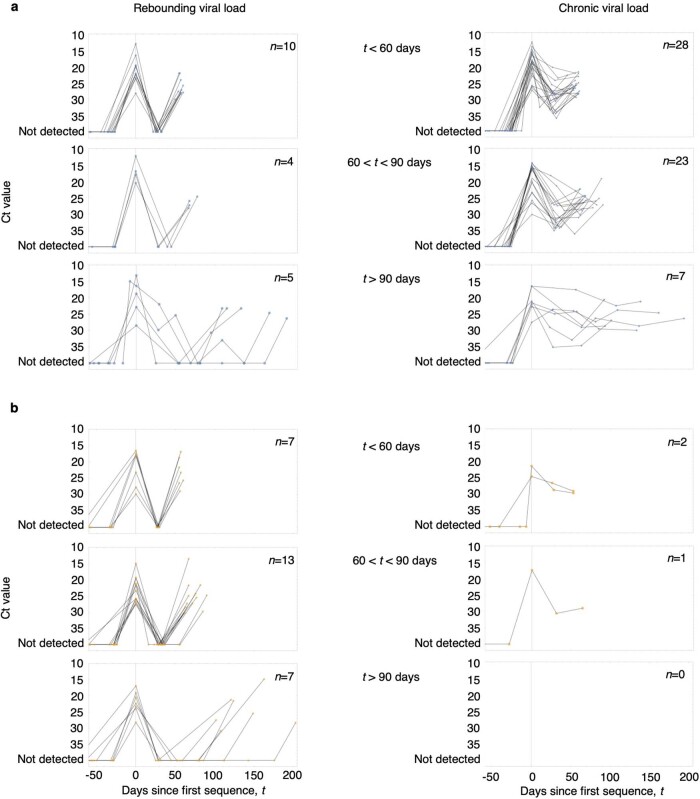
Similar articles
-
Efficacy of late-onset antiviral treatment in immunocompromised hosts with persistent SARS-CoV-2 infection.J Virol. 2024 Sep 17;98(9):e0090524. doi: 10.1128/jvi.00905-24. Epub 2024 Aug 29. J Virol. 2024. PMID: 39207133 Free PMC article.
-
Long-Term Evolution of SARS-CoV-2 in an Immunocompromised Patient with Non-Hodgkin Lymphoma.mSphere. 2021 Aug 25;6(4):e0024421. doi: 10.1128/mSphere.00244-21. Epub 2021 Jul 28. mSphere. 2021. PMID: 34319130 Free PMC article.
-
Early Adoption of Longitudinal Surveillance for SARS-CoV-2 among Staff in Long-Term Care Facilities: Prevalence, Virologic and Sequence Analysis.Microbiol Spectr. 2021 Dec 22;9(3):e0100321. doi: 10.1128/Spectrum.01003-21. Epub 2021 Nov 10. Microbiol Spectr. 2021. PMID: 34756092 Free PMC article.
-
The consequences of SARS-CoV-2 within-host persistence.Nat Rev Microbiol. 2025 May;23(5):288-302. doi: 10.1038/s41579-024-01125-y. Epub 2024 Nov 25. Nat Rev Microbiol. 2025. PMID: 39587352 Review.
-
Intrinsic factors behind long COVID: III. Persistence of SARS-CoV-2 and its components.J Cell Biochem. 2024 Jan;125(1):22-44. doi: 10.1002/jcb.30514. Epub 2023 Dec 14. J Cell Biochem. 2024. PMID: 38098317 Review.
Cited by
-
Factors Associated with Prolonged SARS-CoV-2 Viral Positivity in an Italian Cohort of Hospitalized Patients.Diseases. 2024 Jun 28;12(7):138. doi: 10.3390/diseases12070138. Diseases. 2024. PMID: 39057109 Free PMC article.
-
Respiratory long COVID in aged hamsters features impaired lung function post-exercise with bronchiolization and fibrosis.Nat Commun. 2025 Feb 28;16(1):2080. doi: 10.1038/s41467-025-57267-x. Nat Commun. 2025. PMID: 40021627 Free PMC article.
-
Digestive Manifestations of Post-COVID-19: A Focus on Therapeutic Strategies.Pathogens. 2025 Jun 3;14(6):555. doi: 10.3390/pathogens14060555. Pathogens. 2025. PMID: 40559563 Free PMC article. Review.
-
APOBEC3-related mutations in the spike protein-encoding region facilitate SARS-CoV-2 evolution.Heliyon. 2024 May 29;10(11):e32139. doi: 10.1016/j.heliyon.2024.e32139. eCollection 2024 Jun 15. Heliyon. 2024. PMID: 38868014 Free PMC article.
-
Host-Virus Interface in Persistent SARS-CoV-2 Infections: Viral Characteristic Evolution and Gene Expression Profiling Analysis.Int J Mol Sci. 2025 Jun 27;26(13):6221. doi: 10.3390/ijms26136221. Int J Mol Sci. 2025. PMID: 40649996 Free PMC article.
References
-
- Ghafari, M., Liu, Q., Dhillon, A., Katzourakis, A. & Weissman, D. B. Investigating the evolutionary origins of the first three SARS-CoV-2 variants of concern. Front. Virol.10.3389/fviro.2022.942555 (2022).
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
