

Mechanisms of action and resistance in histone methylation-targeted therapy
- PMID: 38383791
- PMCID: PMC10917674
- DOI: 10.1038/s41586-024-07103-x
Mechanisms of action and resistance in histone methylation-targeted therapy
Abstract
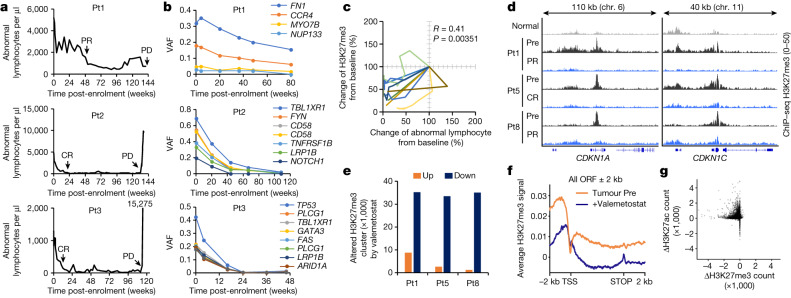
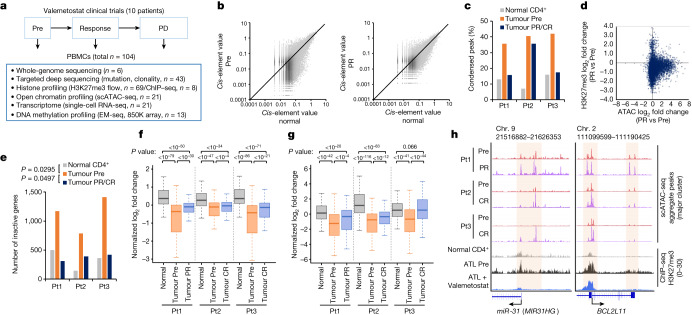
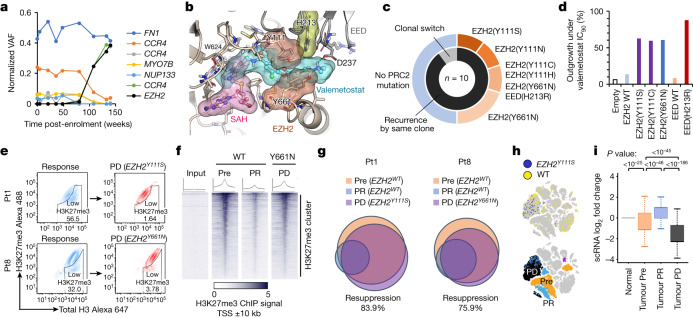
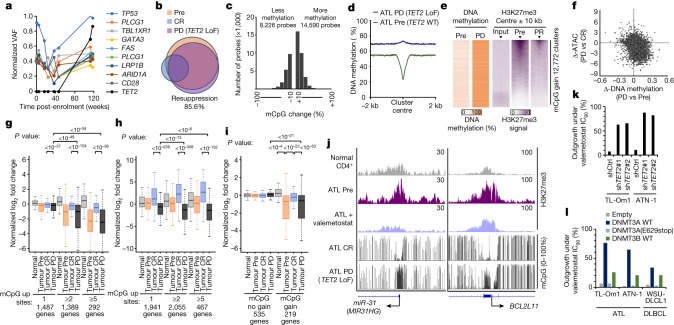
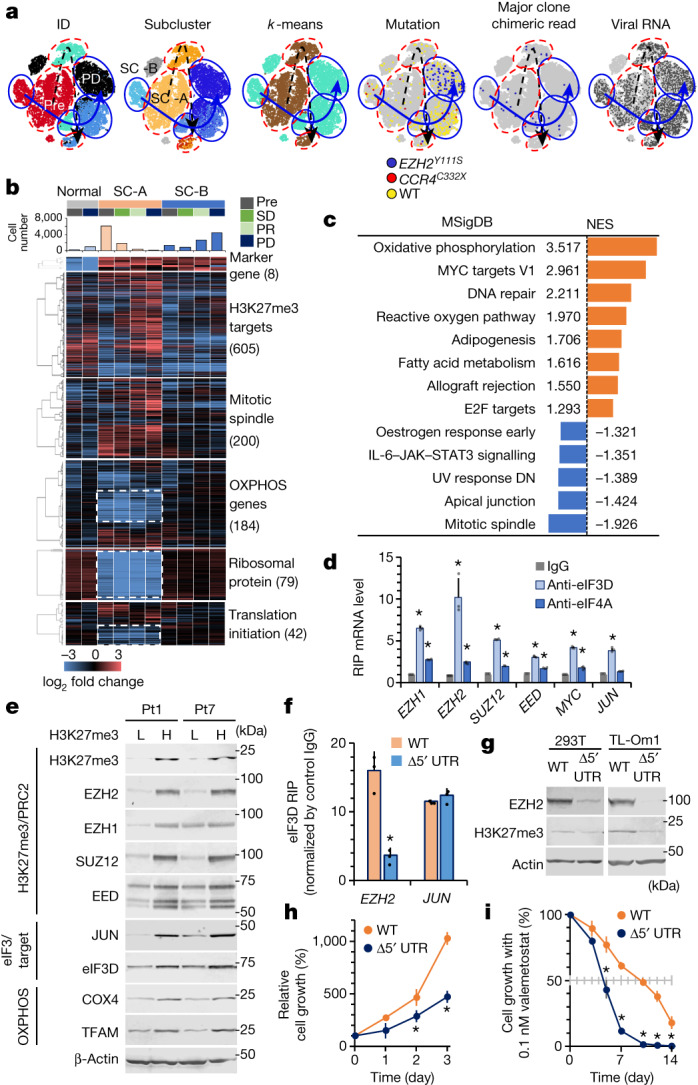
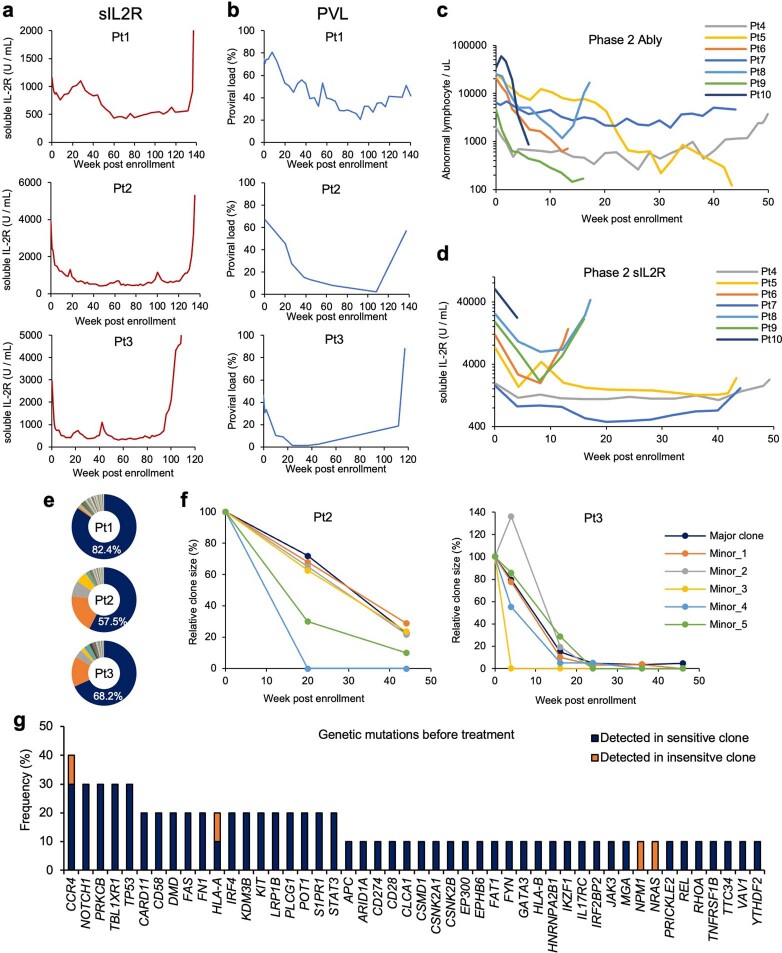
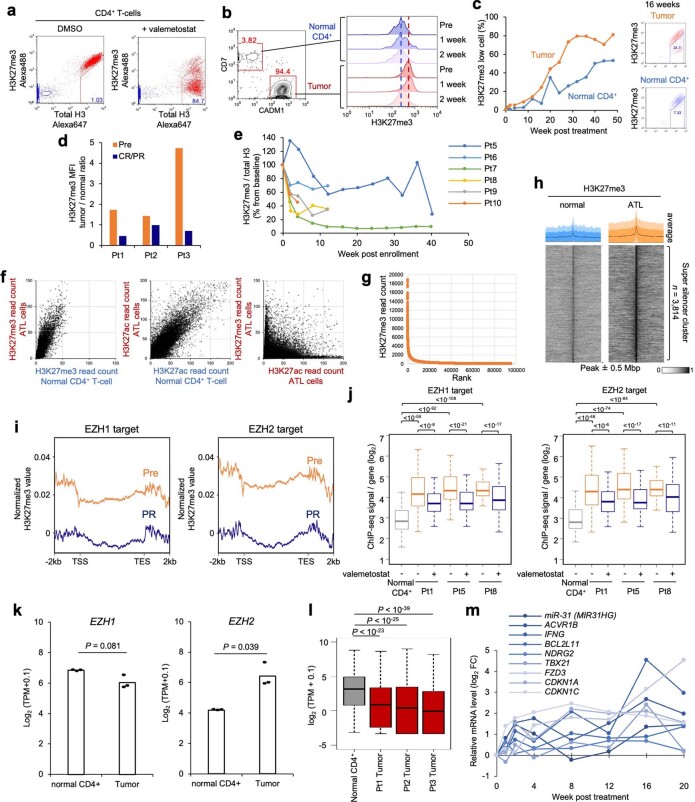
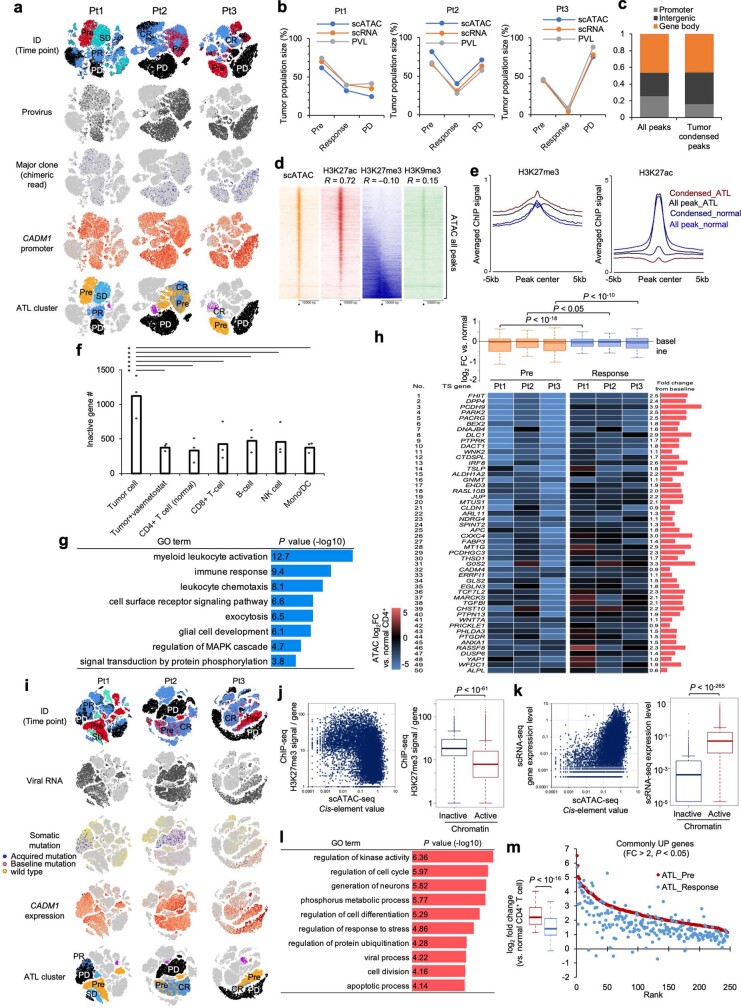
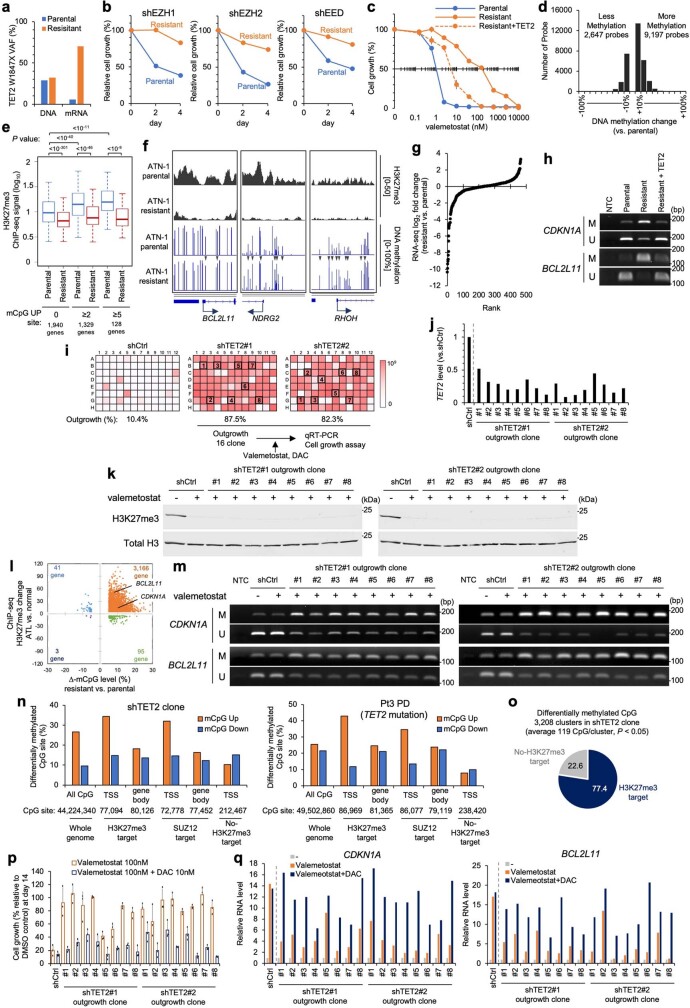
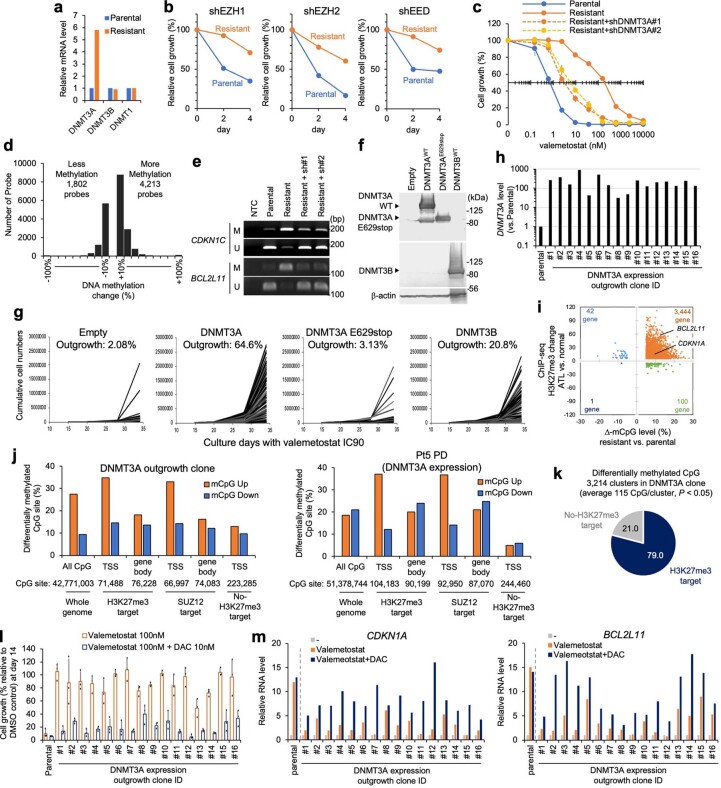
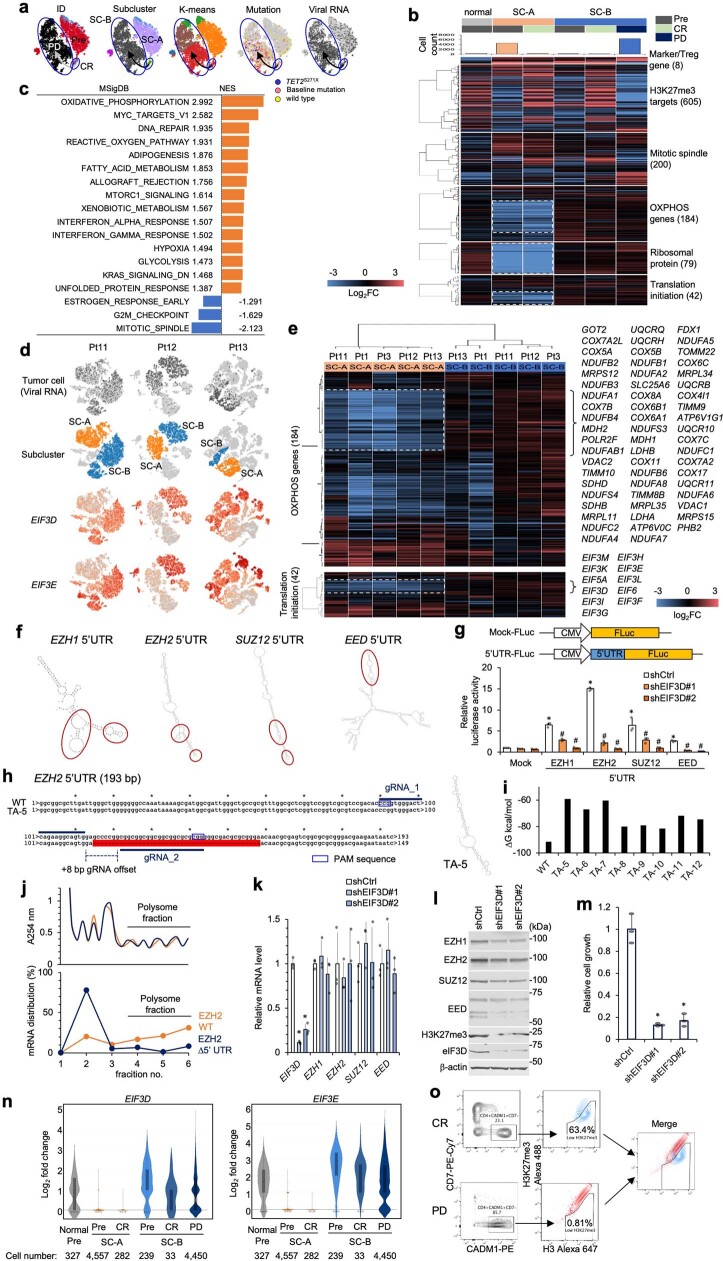
Epigenomes enable the rectification of disordered cancer gene expression, thereby providing new targets for pharmacological interventions. The clinical utility of targeting histone H3 lysine trimethylation (H3K27me3) as an epigenetic hallmark has been demonstrated1-7. However, in actual therapeutic settings, the mechanism by which H3K27me3-targeting therapies exert their effects and the response of tumour cells remain unclear. Here we show the potency and mechanisms of action and resistance of the EZH1-EZH2 dual inhibitor valemetostat in clinical trials of patients with adult T cell leukaemia/lymphoma. Administration of valemetostat reduced tumour size and demonstrated durable clinical response in aggressive lymphomas with multiple genetic mutations. Integrative single-cell analyses showed that valemetostat abolishes the highly condensed chromatin structure formed by the plastic H3K27me3 and neutralizes multiple gene loci, including tumour suppressor genes. Nevertheless, subsequent long-term treatment encounters the emergence of resistant clones with reconstructed aggregate chromatin that closely resemble the pre-dose state. Acquired mutations at the PRC2-compound interface result in the propagation of clones with increased H3K27me3 expression. In patients free of PRC2 mutations, TET2 mutation or elevated DNMT3A expression causes similar chromatin recondensation through de novo DNA methylation in the H3K27me3-associated regions. We identified subpopulations with distinct metabolic and gene translation characteristics implicated in primary susceptibility until the acquisition of the heritable (epi)mutations. Targeting epigenetic drivers and chromatin homeostasis may provide opportunities for further sustained epigenetic cancer therapies.
© 2024. The Author(s).
Conflict of interest statement
M.Y., Y.S. and K.U. received research funding from Daiichi Sankyo Co., Ltd. I.Y. is an employee of Daiichi Sankyo RD Novare Co., Ltd. G.T., Y.H., K.Ito and S.T. are employees of Daiichi Sankyo Co., Ltd. Daiichi Sankyo Co., Ltd holds substance patents on valemetostat. The University of Tokyo and Daiichi Sankyo Co., Ltd hold patents for application of valemetostat to patients with ATL and individuals infected with HTLV-1. M.Y. and T.W. are named as inventors. All other authors declare no competing interests.
Figures
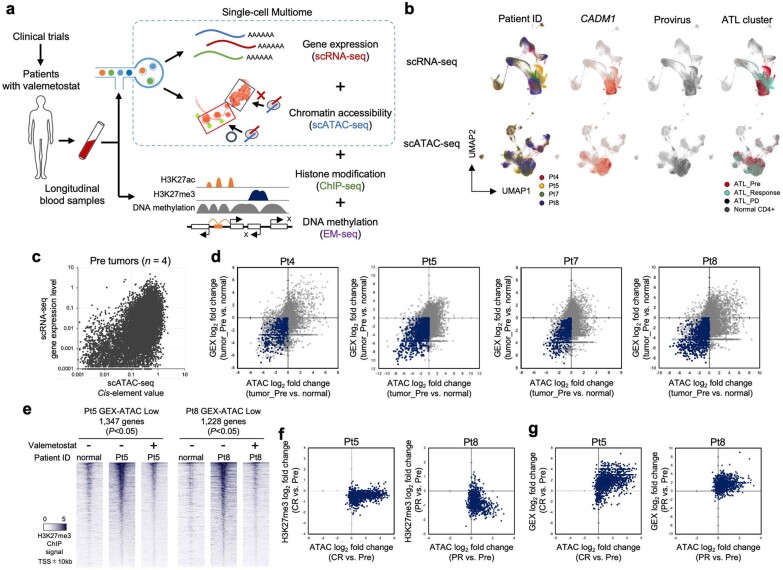
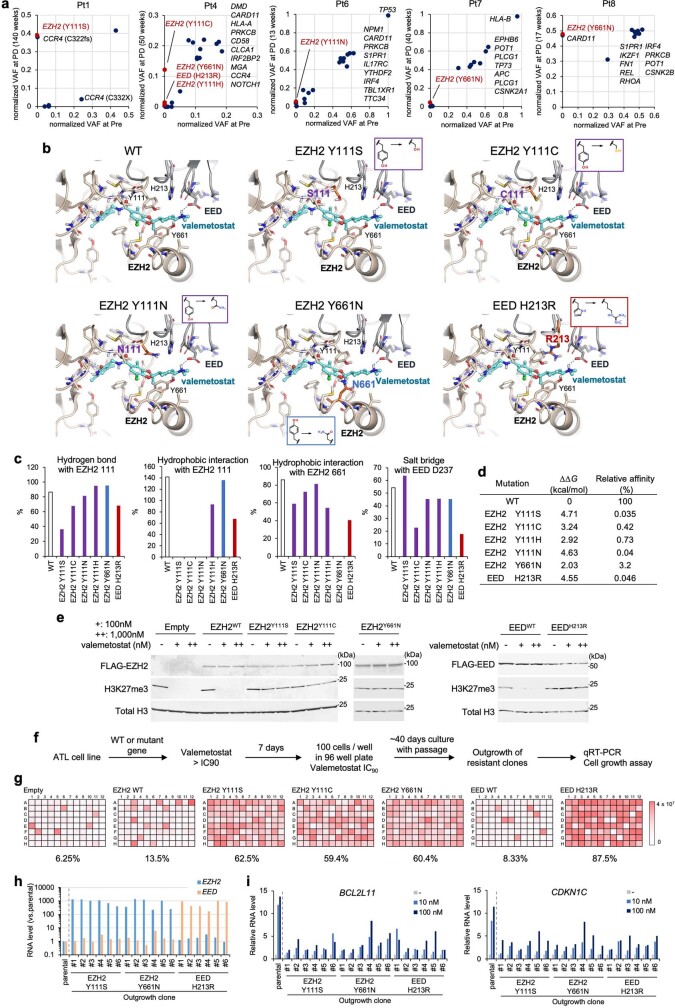
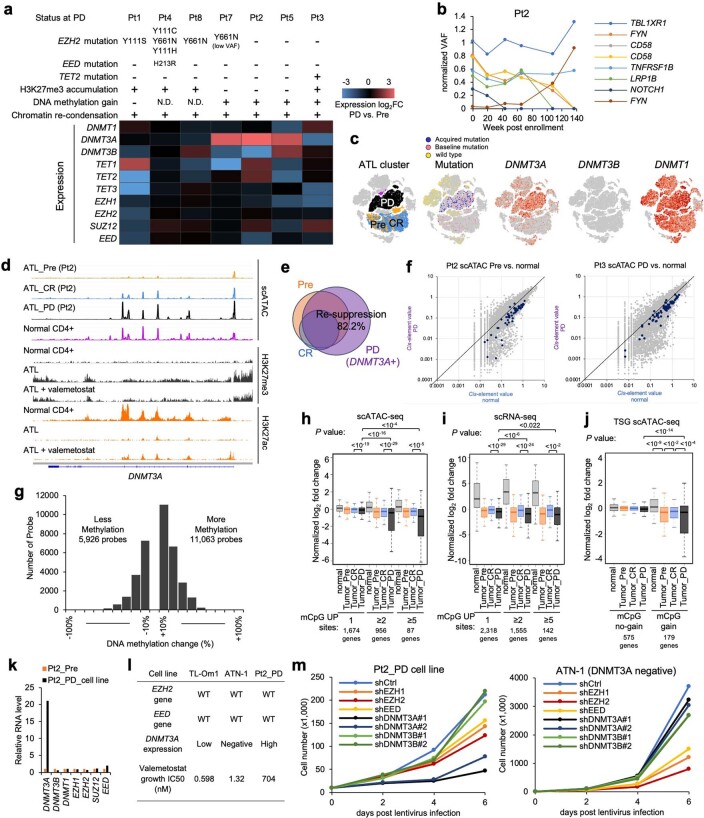
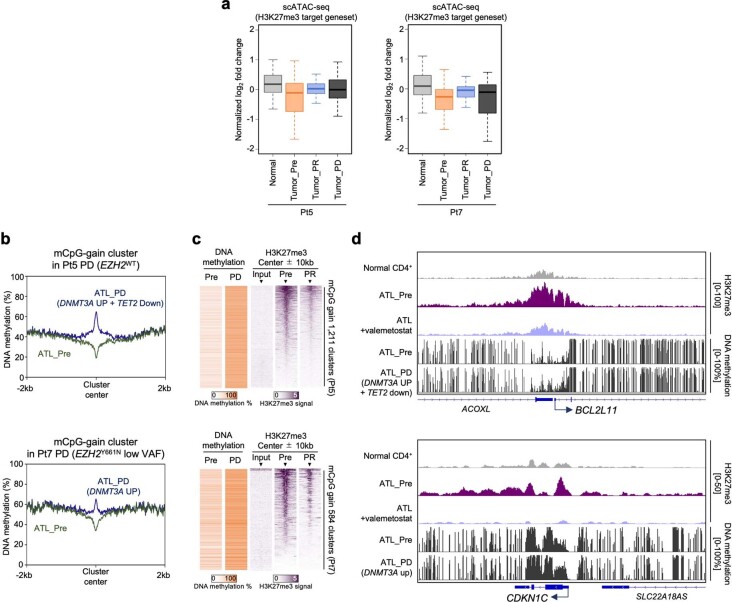
References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
