

Acquired Cross-Resistance in Small Cell Lung Cancer due to Extrachromosomal DNA Amplification of MYC Paralogs
- PMID: 38386926
- PMCID: PMC11061613
- DOI: 10.1158/2159-8290.CD-23-0656
Acquired Cross-Resistance in Small Cell Lung Cancer due to Extrachromosomal DNA Amplification of MYC Paralogs
Abstract
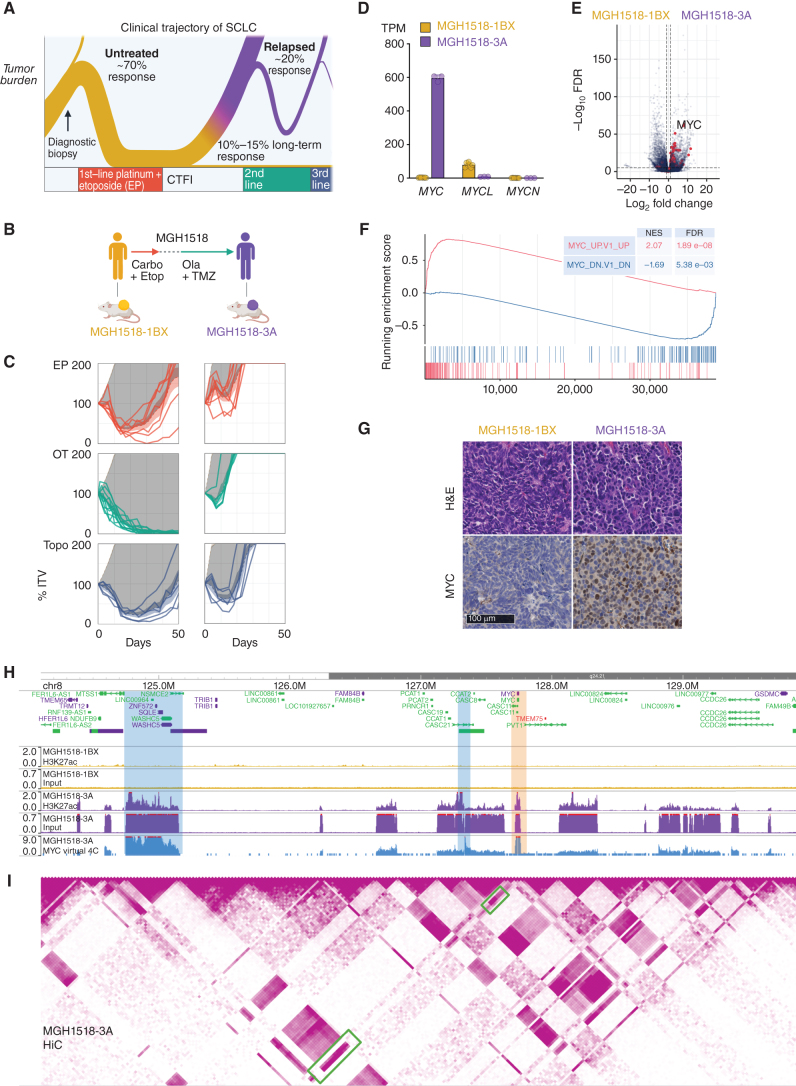
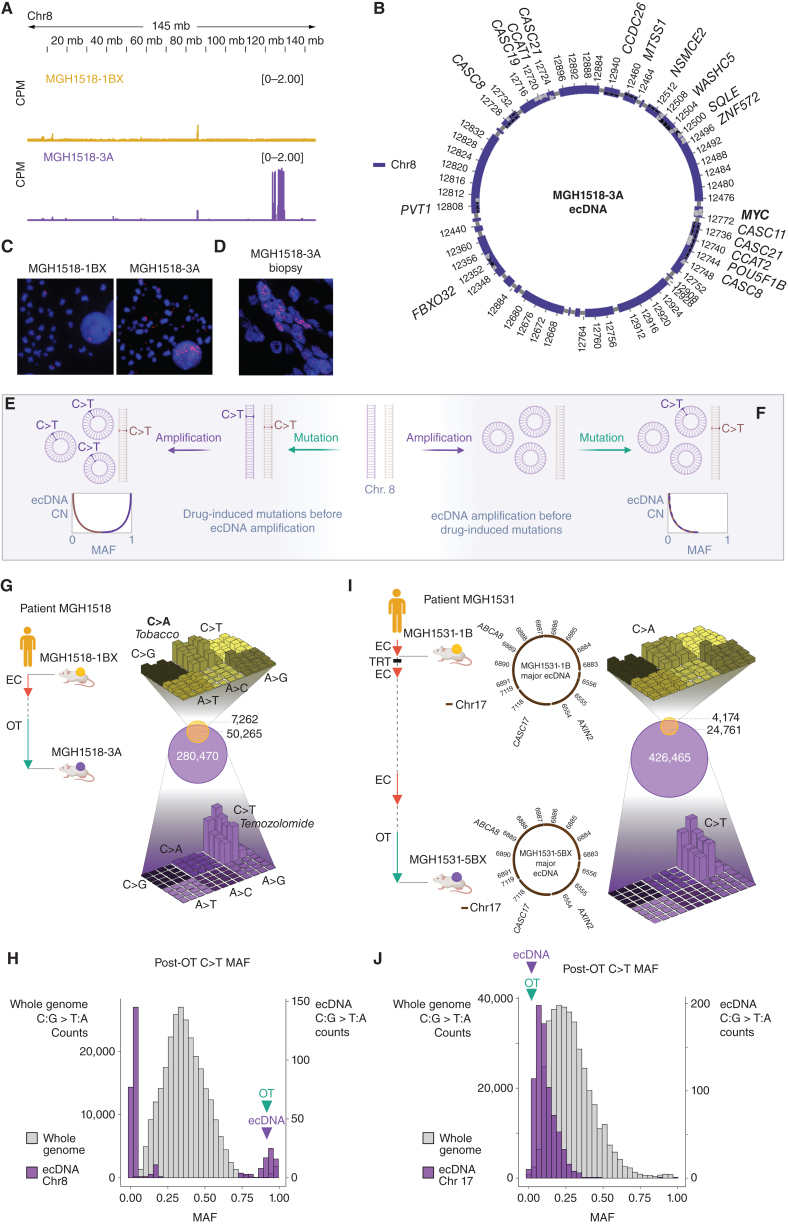
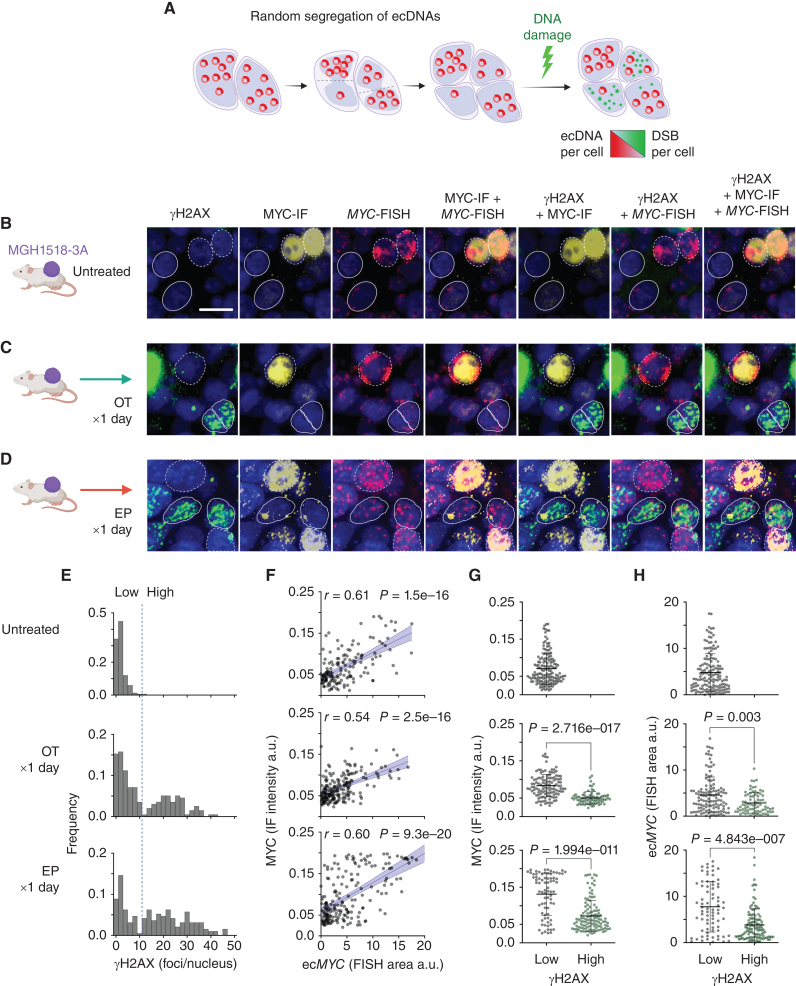
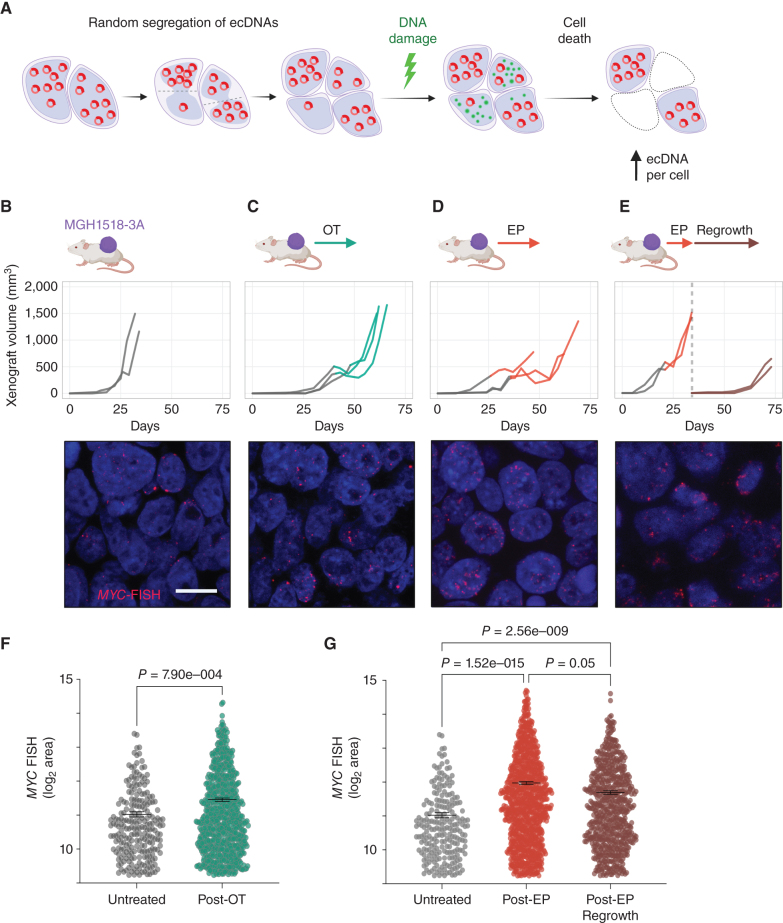
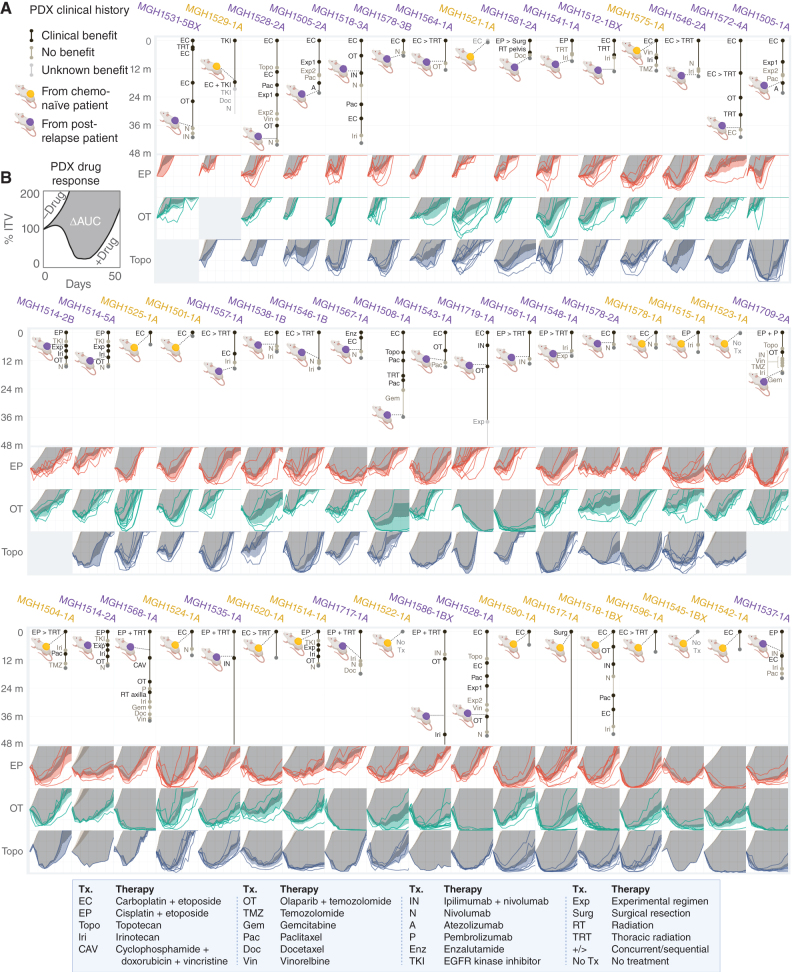
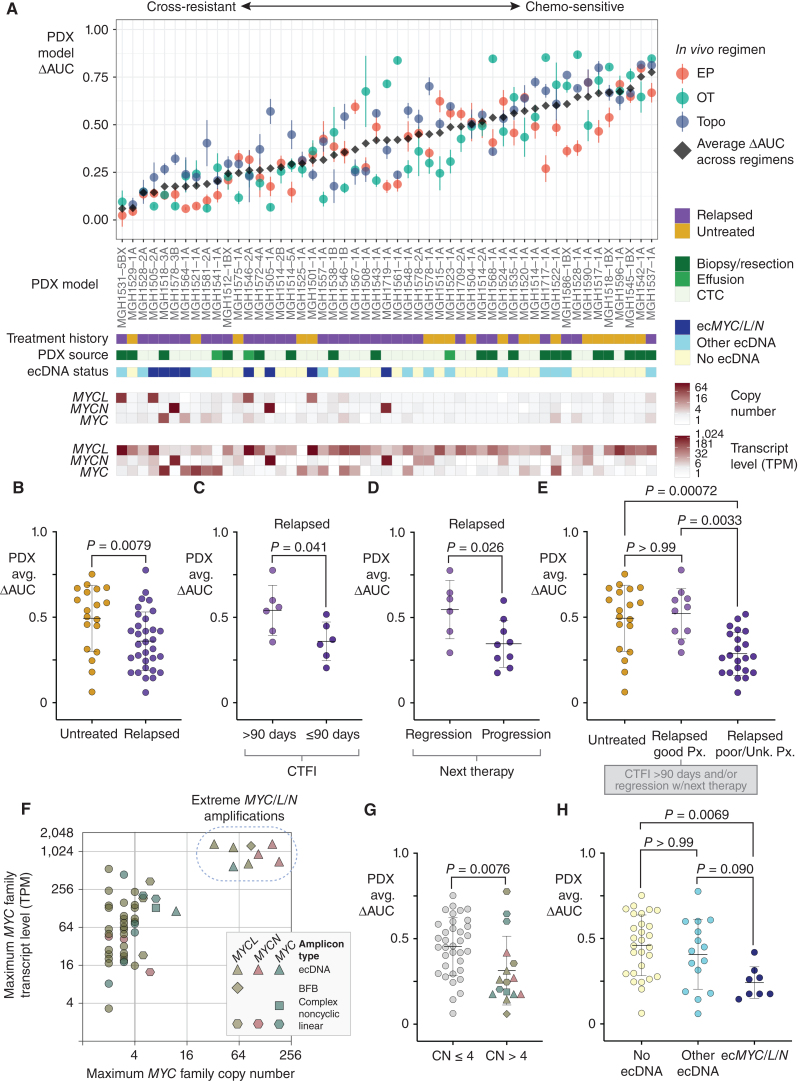
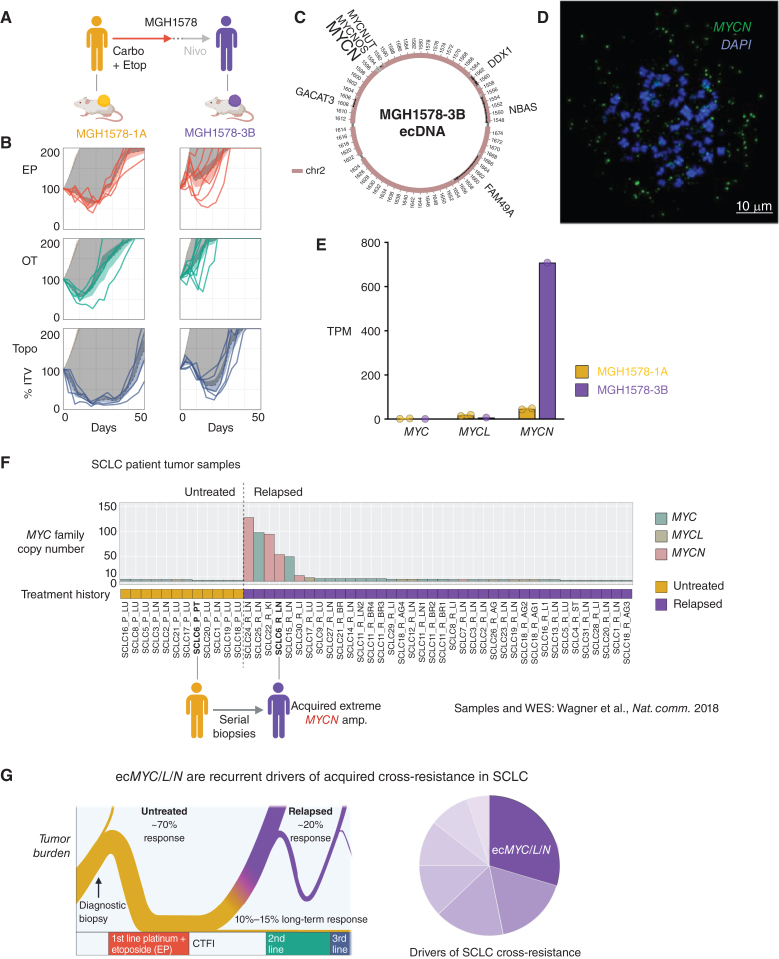
Small cell lung cancer (SCLC) presents as a highly chemosensitive malignancy but acquires cross-resistance after relapse. This transformation is nearly inevitable in patients but has been difficult to capture in laboratory models. Here, we present a preclinical system that recapitulates acquired cross-resistance, developed from 51 patient-derived xenograft (PDX) models. Each model was tested in vivo against three clinical regimens: cisplatin plus etoposide, olaparib plus temozolomide, and topotecan. These drug-response profiles captured hallmark clinical features of SCLC, such as the emergence of treatment-refractory disease after early relapse. For one patient, serial PDX models revealed that cross-resistance was acquired through MYC amplification on extrachromosomal DNA (ecDNA). Genomic and transcriptional profiles of the full PDX panel revealed that MYC paralog amplifications on ecDNAs were recurrent in relapsed cross-resistant SCLC, and this was corroborated in tumor biopsies from relapsed patients. We conclude that ecDNAs with MYC paralogs are recurrent drivers of cross-resistance in SCLC.
Significance: SCLC is initially chemosensitive, but acquired cross-resistance renders this disease refractory to further treatment and ultimately fatal. The genomic drivers of this transformation are unknown. We use a population of PDX models to discover that amplifications of MYC paralogs on ecDNA are recurrent drivers of acquired cross-resistance in SCLC. This article is featured in Selected Articles from This Issue, p. 695.
©2024 The Authors; Published by the American Association for Cancer Research.
Figures
Update of
-
Acquired Cross-resistance in Small Cell Lung Cancer due to Extrachromosomal DNA Amplification of MYC paralogs.bioRxiv [Preprint]. 2023 Jun 28:2023.06.23.546278. doi: 10.1101/2023.06.23.546278. bioRxiv. 2023. Update in: Cancer Discov. 2024 May 1;14(5):804-827. doi: 10.1158/2159-8290.CD-23-0656. PMID: 37425738 Free PMC article. Updated. Preprint.
References
-
- Siegel RL, Miller KD, Wagle NS, Jemal A. Cancer statistics, 2023. CA Cancer J Clin 2023;73:17–48. - PubMed
-
- Bray F, Colombet M, Mery L, Piñeros M, Znaor A, Zanetti R. Cancer incidence in five continents. Volume XI. IARC Scientific Publication No. 166; 2021.
-
- Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. . Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin 2021;71:209–49. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
