

Chromosome evolution screens recapitulate tissue-specific tumor aneuploidy patterns
- PMID: 38388848
- PMCID: PMC11096114
- DOI: 10.1038/s41588-024-01665-2
Chromosome evolution screens recapitulate tissue-specific tumor aneuploidy patterns
Abstract
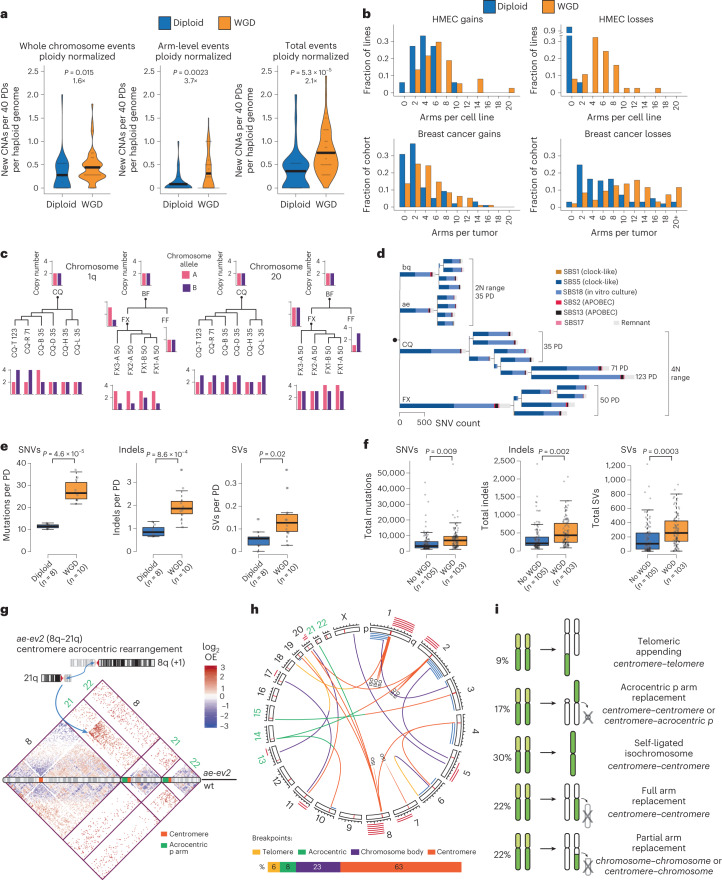
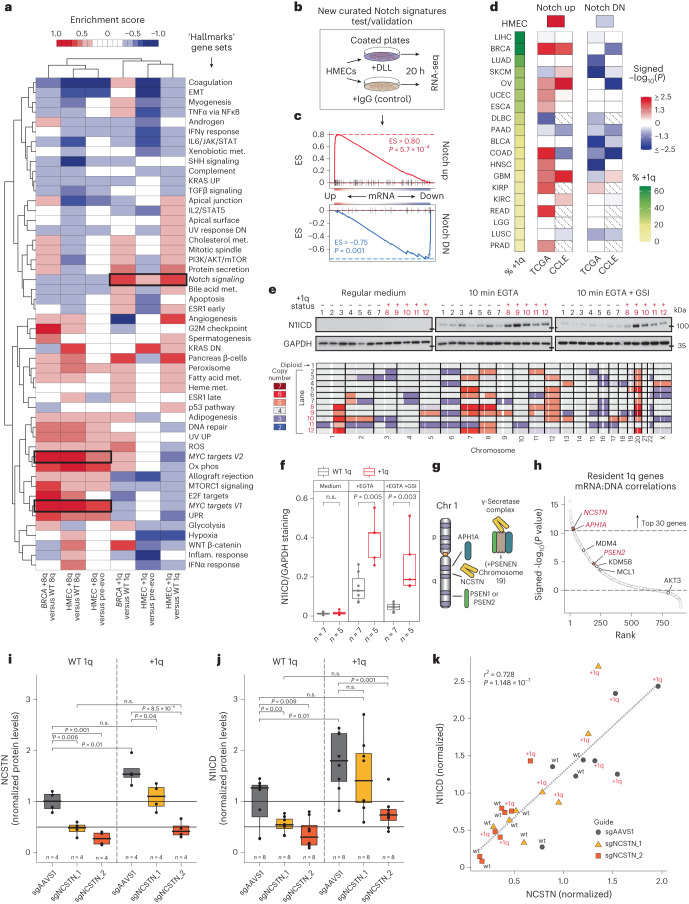
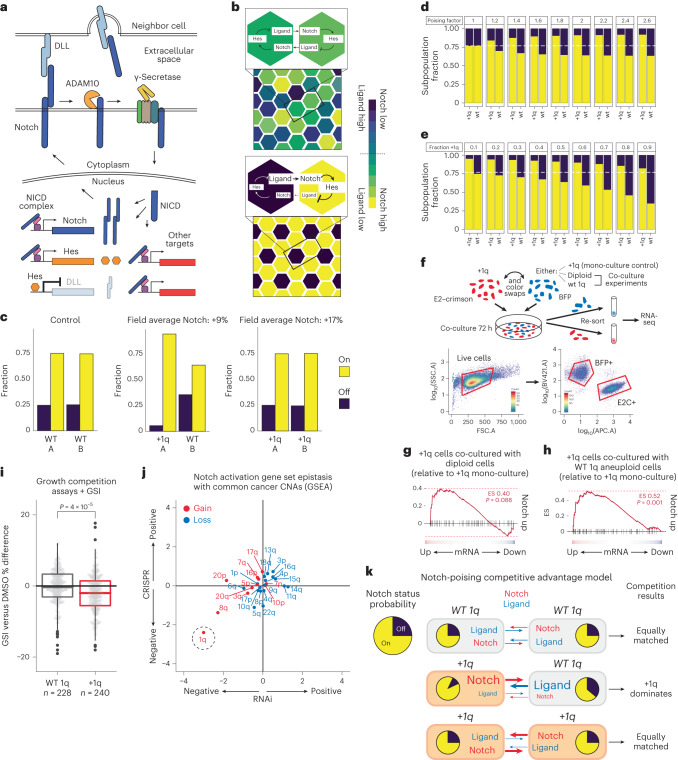
Whole chromosome and arm-level copy number alterations occur at high frequencies in tumors, but their selective advantages, if any, are poorly understood. Here, utilizing unbiased whole chromosome genetic screens combined with in vitro evolution to generate arm- and subarm-level events, we iteratively selected the fittest karyotypes from aneuploidized human renal and mammary epithelial cells. Proliferation-based karyotype selection in these epithelial lines modeled tissue-specific tumor aneuploidy patterns in patient cohorts in the absence of driver mutations. Hi-C-based translocation mapping revealed that arm-level events usually emerged in multiples of two via centromeric translocations and occurred more frequently in tetraploids than diploids, contributing to the increased diversity in evolving tetraploid populations. Isogenic clonal lineages enabled elucidation of pro-tumorigenic mechanisms associated with common copy number alterations, revealing Notch signaling potentiation as a driver of 1q gain in breast cancer. We propose that intrinsic, tissue-specific proliferative effects underlie tumor copy number patterns in cancer.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures













References
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
