

CAGI, the Critical Assessment of Genome Interpretation, establishes progress and prospects for computational genetic variant interpretation methods
- PMID: 38389099
- PMCID: PMC10882881
- DOI: 10.1186/s13059-023-03113-6
CAGI, the Critical Assessment of Genome Interpretation, establishes progress and prospects for computational genetic variant interpretation methods
Abstract
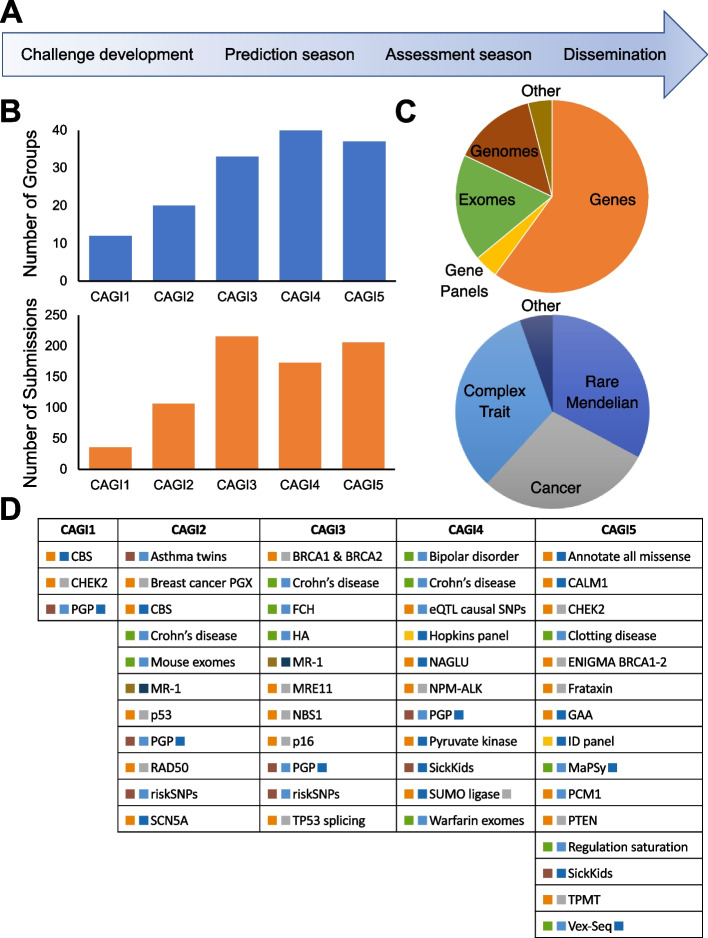
Background: The Critical Assessment of Genome Interpretation (CAGI) aims to advance the state-of-the-art for computational prediction of genetic variant impact, particularly where relevant to disease. The five complete editions of the CAGI community experiment comprised 50 challenges, in which participants made blind predictions of phenotypes from genetic data, and these were evaluated by independent assessors.
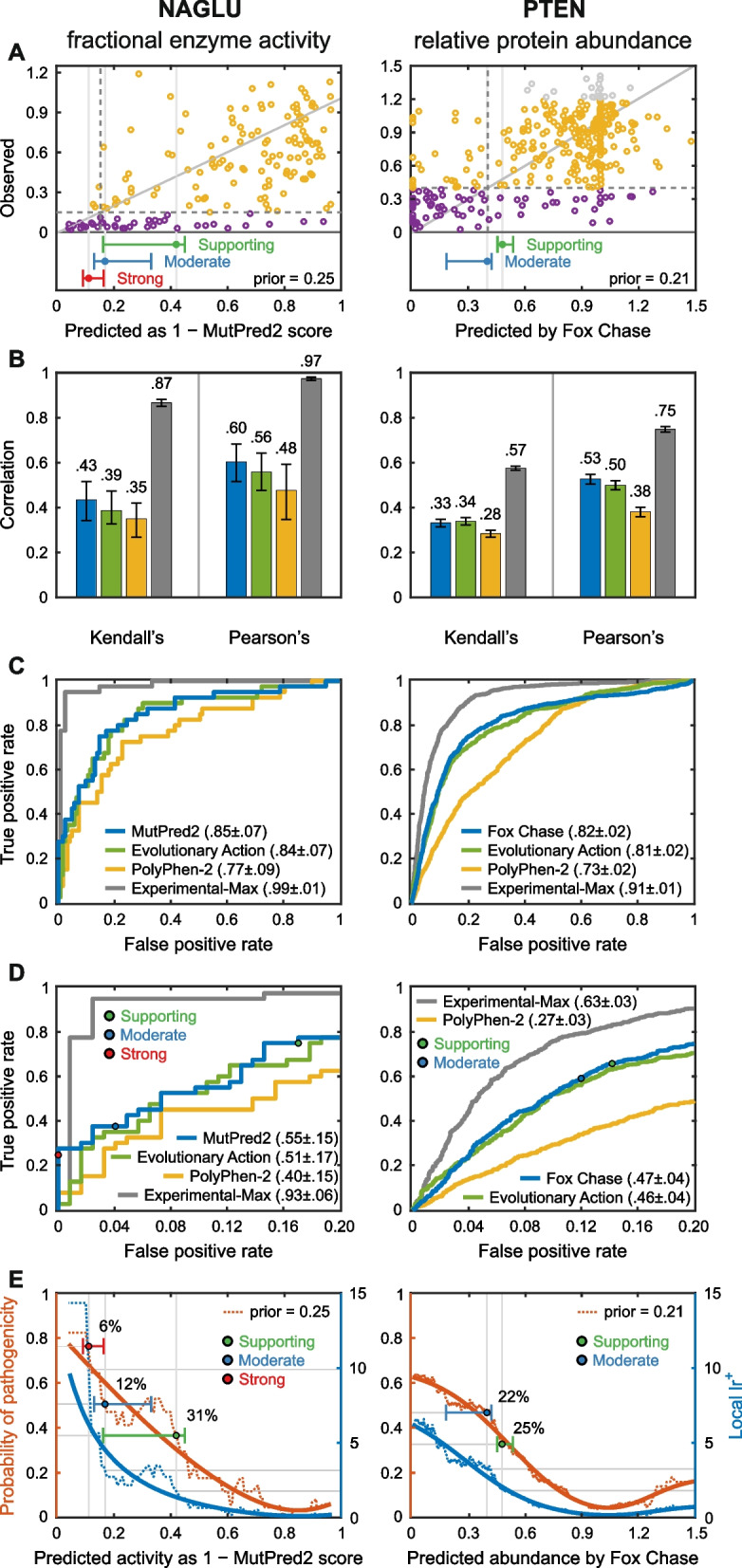
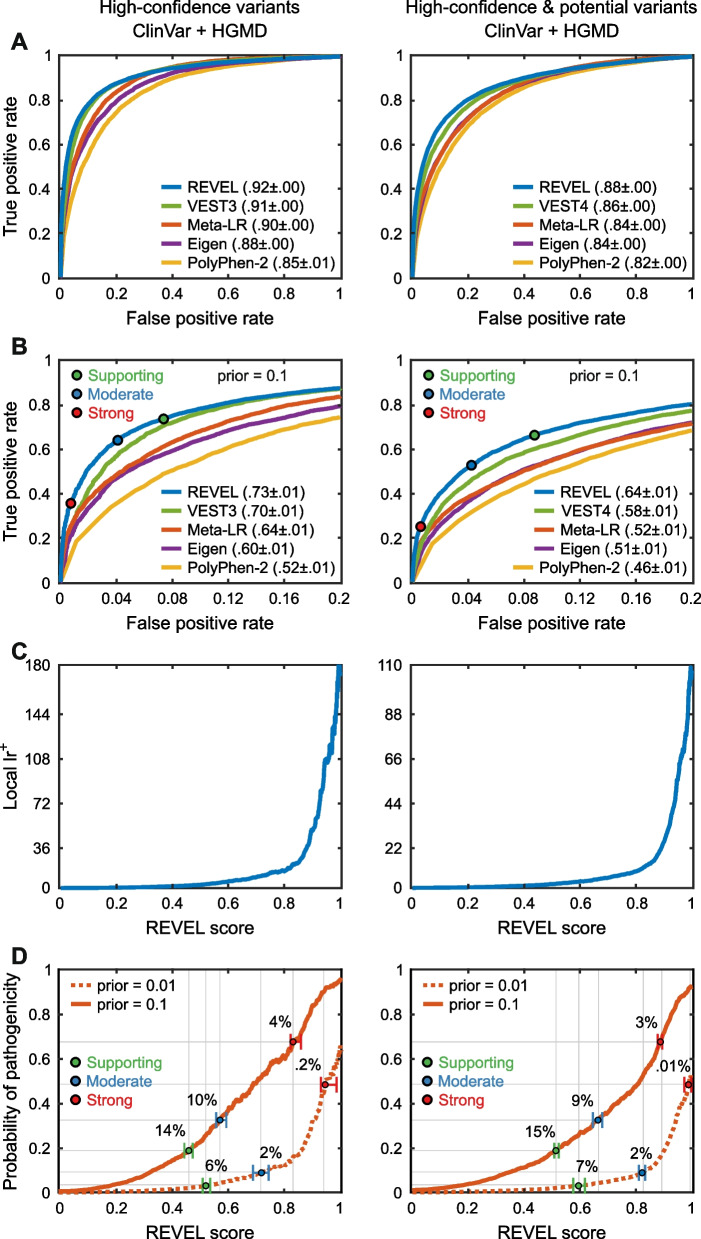
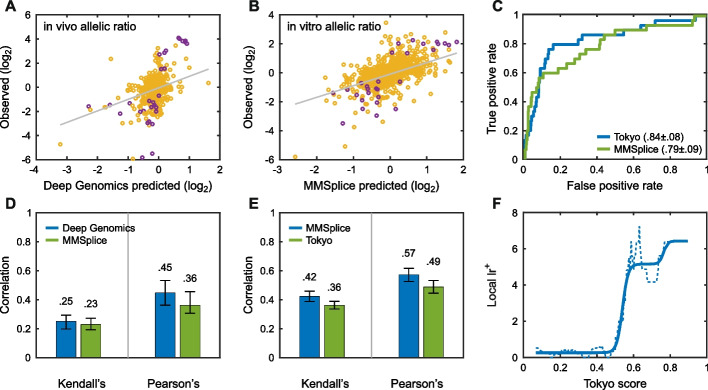
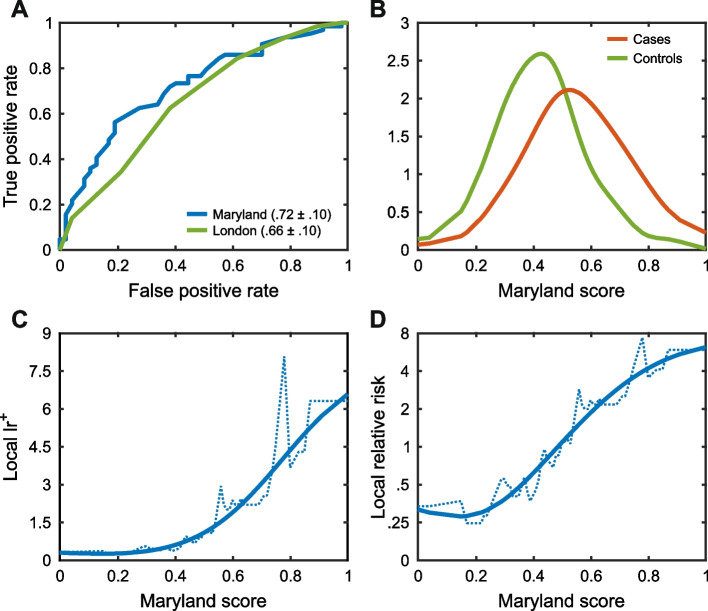
Results: Performance was particularly strong for clinical pathogenic variants, including some difficult-to-diagnose cases, and extends to interpretation of cancer-related variants. Missense variant interpretation methods were able to estimate biochemical effects with increasing accuracy. Assessment of methods for regulatory variants and complex trait disease risk was less definitive and indicates performance potentially suitable for auxiliary use in the clinic.
Conclusions: Results show that while current methods are imperfect, they have major utility for research and clinical applications. Emerging methods and increasingly large, robust datasets for training and assessment promise further progress ahead.
© 2024. The Author(s).
Conflict of interest statement
Principal authors of this paper participated as predictors in many of the CAGI challenges reported. The unified numerical framework employed for reanalysis of the challenges yields results that are consistent with those obtained by the independent assessors of each challenge and in particular selected methods are the highest ranked in the independent assessments. Nevertheless, while every care was taken to mitigate any potential biases in this work, the authors’ participation in CAGI may have affected the presentation of findings, including the selection of challenges, metrics, assessment criteria, and emphasis given on particular results.
VBG is a current employee and shareholder of AstraZeneca; RB is a shareholder of enGenome; AJB is a co-founder and consultant to Personalis and NuMedii as well as a consultant to Samsung, Mango Tree Corporation and in the recent past, 10 × Genomics, Helix and Pathway; Carles Corbi-Verge is a computational scientist at the drug discovery company; Cyclica INC and is compensated with income and equity; KC is one of the Regeneron authors and owns options and/or stock of the company; DD is Chief Scientist at Geneyx Genomex Ltd; CD is a consultant to Exact Sciences and is compensated with income and equity; GAG receives research funds from IBM and Pharmacyclics and is an inventor on patent applications related to MSMuTect, MSMutSig, MSIDetect, POLYSOLVER, and SignatureAnalyzer-GPU, and is a founder, consultant, and holds privately held equity in Scorpion Therapeutics; NG is an employee and stockholder at Pacific Biosciences; RH is a paid consultant for Invitae and Scientific Advisory Board member for Variant Bio; AK is a consultant at Illumina Inc., Scientific Advisory Board member of OpenTargets; KK is one of the Regeneron authors and owns options and/or stock of the company; IL is an employer and stockholder of enGenome; MSM owns stock in PhenoTips; GN is an employee of enGenome; AOD-L is a member of the Scientific Advisory Board of Congenica; ER is a shareholder of enGenome; PKR is the founder of CytoGnomix; FPR is a shareholder in Ranomics and SeqWell, an advisor for SeqWell, BioSymetrics, and Constantiam BioSciences, and has received research sponsorships from Biogen, Alnylam, Deep Genomics, and Beam Therapeutics; PCS is the co-founder and shareholder of Sherlock Biosciences, a board member and shareholder of Danaher Corporation, and has filed patents related to this work; PLFT is an employer and stockholder in AccuraGen; RT has filed patents related to this work; MHW is a shareholder of Beth Bioinformatics Co., Ltd.; CMY is an employee and shareholder of Vertex Pharmaceuticals; JZ is an employee of AstraZeneca; SEB receives support at the University of California, Berkeley from a research agreement from TCS.
Figures

Comment on
-
Reports from CAGI: The Critical Assessment of Genome Interpretation.Hum Mutat. 2017 Sep;38(9):1039-1041. doi: 10.1002/humu.23290. Hum Mutat. 2017. PMID: 28817245 Free PMC article. No abstract available.
-
Reports from the fifth edition of CAGI: The Critical Assessment of Genome Interpretation.Hum Mutat. 2019 Sep;40(9):1197-1201. doi: 10.1002/humu.23876. Epub 2019 Aug 26. Hum Mutat. 2019. PMID: 31334884 Free PMC article.
References
-
- Nielsen FC, van Overeem HT, Sorensen CS. Hereditary breast and ovarian cancer: new genes in confined pathways. Nat Rev Cancer. 2016;16(9):599–612. - PubMed
Publication types
MeSH terms
Grants and funding
- U24 HG010615/HG/NHGRI NIH HHS/United States
- R01 HD103805/HD/NICHD NIH HHS/United States
- U01 HG012022/HG/NHGRI NIH HHS/United States
- R35 GM127131/GM/NIGMS NIH HHS/United States
- R01 GM102365/GM/NIGMS NIH HHS/United States
- U01 AG068214/AG/NIA NIH HHS/United States
- R35 HG011329/HG/NHGRI NIH HHS/United States
- U24 HG007234/HG/NHGRI NIH HHS/United States
- R35 GM124952/GM/NIGMS NIH HHS/United States
- U24 HG009293/HG/NHGRI NIH HHS/United States
- R01 GM109146/GM/NIGMS NIH HHS/United States
- U41 HG007346/HG/NHGRI NIH HHS/United States
- R01 HG010372/HG/NHGRI NIH HHS/United States
- U41 HG009293/HG/NHGRI NIH HHS/United States
- R01 AG074009/AG/NIA NIH HHS/United States
- R35 GM118140/GM/NIGMS NIH HHS/United States
- U24 HG007346/HG/NHGRI NIH HHS/United States
- R00 LM012992/LM/NLM NIH HHS/United States
- U41 HG007234/HG/NHGRI NIH HHS/United States
- R35 GM130361/GM/NIGMS NIH HHS/United States
- R24 OD011883/OD/NIH HHS/United States
- U01 HG009380/HG/NHGRI NIH HHS/United States
- R01 GM078598/GM/NIGMS NIH HHS/United States
- P30 CA045508/CA/NCI NIH HHS/United States
- R13 HG006650/HG/NHGRI NIH HHS/United States
- R35 GM122517/GM/NIGMS NIH HHS/United States
- R01 CA264971/CA/NCI NIH HHS/United States
- U19 HD077627/HD/NICHD NIH HHS/United States
- RM1 HG010860/HG/NHGRI NIH HHS/United States
- 098051/WT_/Wellcome Trust/United Kingdom
