

Time-series analysis of hematopoietic stem cells
- PMID: 38394540
- PMCID: PMC11309688
- DOI: 10.1097/MD.0000000000036509
Time-series analysis of hematopoietic stem cells
Abstract
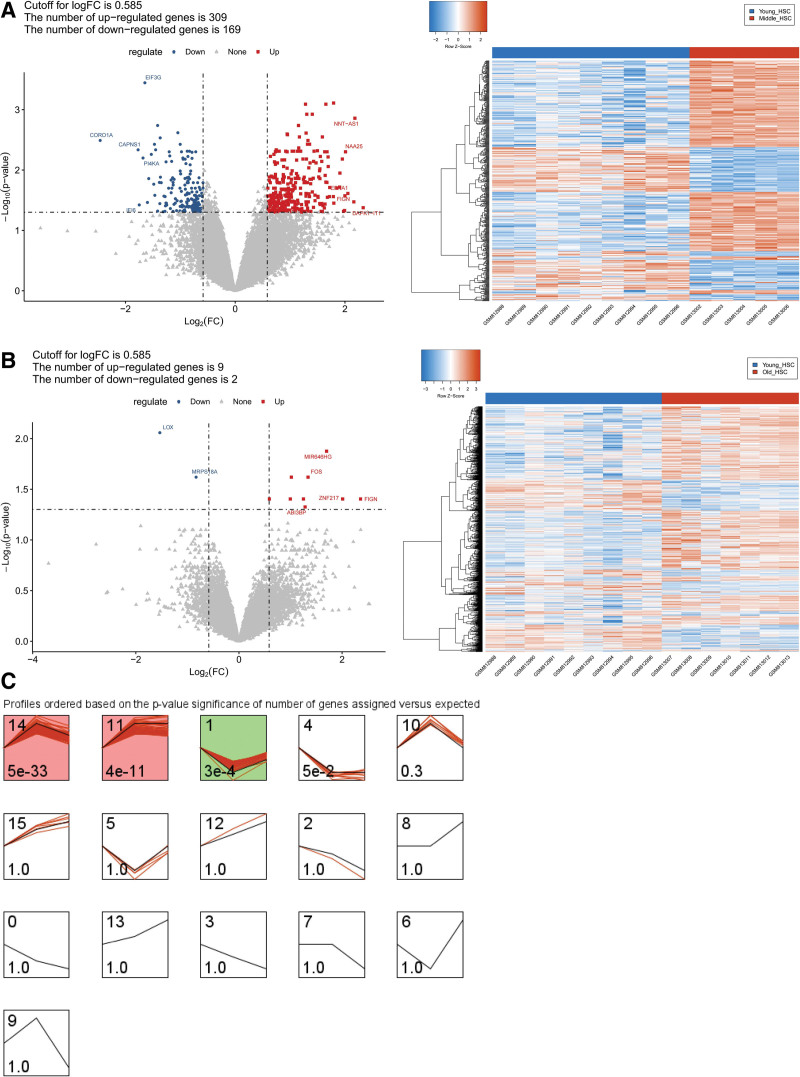
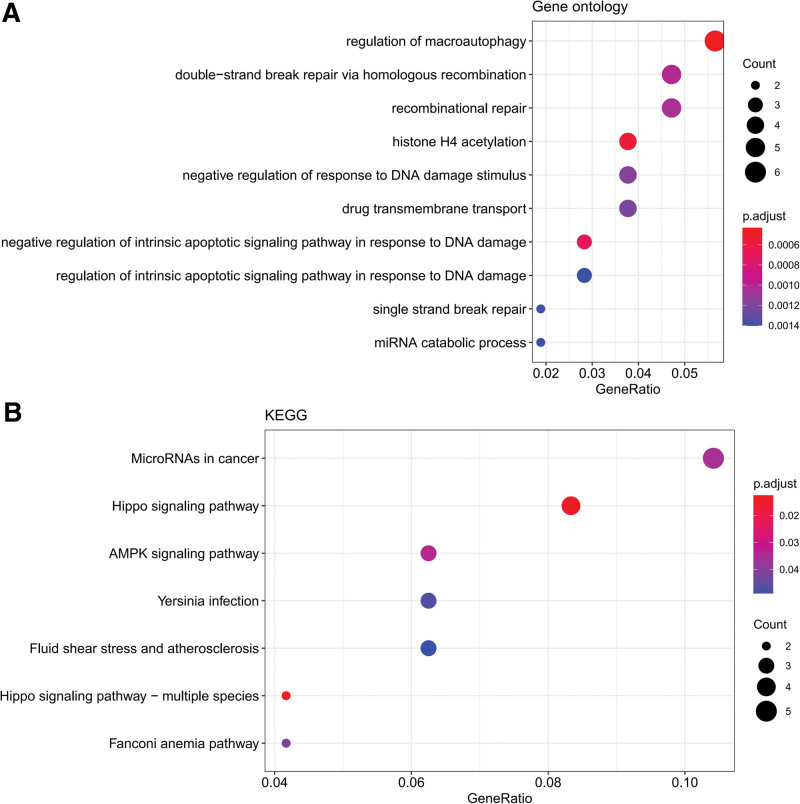
This study aimed to investigate the molecular mechanisms underlying the aging of hematopoietic stem cells (HSCs). Gene expression profile GSE32719 was downloaded from the Gene Expression Omnibus database, including 14 young, 5 middle, and 8 old HSCs. Differential expression analysis, short time-series expression miner analysis, and weighted co-expression network analysis were conducted to screen for hub genes whose expression changed over time during HSC aging. Subsequently, functional enrichment and multiple regulatory network analyses of the hub genes were performed. A total of 124 intersecting time-dependent differentially expressed and module genes were obtained, which were considered hub genes whose expression changed over time during HSC aging. Hub genes were significantly enriched in pathways such as the Hippo and AMP-activated protein kinase (AMPK) signaling pathways. Moreover, AP-1 Transcription Factor Subunit (FOS) and sirtuin 1 (SIRT1) had higher degrees in the protein-protein interaction network, were regulated by more transcription factors (TFs), such as Sp1 transcription factor (SP1) and BRCA1 DNA repair-associated (BRCA1), in the TF-mRNA-miRNA network, were associated with more diseases in the disease-gene network, and could be targeted by more drugs in the drug-gene network. Furthermore, SIRT1 was targeted by miR-9-5p in the TF-mRNA-miRNA network. Hub genes such as FOS and SIRT1 and key pathways such as the Hippo and AMPK signaling pathways may play crucial roles in HSC aging. Moreover, FOS and SIRT1 were regulated by SP1 and BRCA1, respectively, during HSC aging. Furthermore, miR-9-5p may modulate HSC aging by targeting SIRT1. Thus, FOS and SIRT1 may be potential therapeutic targets for age-related hematopoietic dysfunction.
Copyright © 2024 the Author(s). Published by Wolters Kluwer Health, Inc.
Conflict of interest statement
The authors have no conflicts of interest to disclose.
Figures



Similar articles
-
Identification of Serum Exosome-Derived circRNA-miRNA-TF-mRNA Regulatory Network in Postmenopausal Osteoporosis Using Bioinformatics Analysis and Validation in Peripheral Blood-Derived Mononuclear Cells.Front Endocrinol (Lausanne). 2022 Jun 9;13:899503. doi: 10.3389/fendo.2022.899503. eCollection 2022. Front Endocrinol (Lausanne). 2022. PMID: 35757392 Free PMC article.
-
An integrated network analysis approach to identify potential key genes, transcription factors, and microRNAs regulating human hematopoietic stem cell aging.Mol Omics. 2021 Dec 6;17(6):967-984. doi: 10.1039/d1mo00199j. Mol Omics. 2021. PMID: 34605522
-
Integrated miRNA-mRNA network revealing the key molecular characteristics of ossification of the posterior longitudinal ligament.Medicine (Baltimore). 2020 May 22;99(21):e20268. doi: 10.1097/MD.0000000000020268. Medicine (Baltimore). 2020. PMID: 32481304 Free PMC article.
-
Identification of microRNA-mRNA-TF regulatory networks in periodontitis by bioinformatics analysis.BMC Oral Health. 2022 Apr 9;22(1):118. doi: 10.1186/s12903-022-02150-0. BMC Oral Health. 2022. PMID: 35397550 Free PMC article.
-
Integrated analysis of competing endogenous RNA (ceRNA) networks in subacute stage of spinal cord injury.Gene. 2020 Feb 5;726:144171. doi: 10.1016/j.gene.2019.144171. Epub 2019 Oct 26. Gene. 2020. PMID: 31669638
References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous
