Robustness of cancer microbiome signals over a broad range of methodological variation
- PMID: 38396294
- PMCID: PMC10997506
- DOI: 10.1038/s41388-024-02974-w
Robustness of cancer microbiome signals over a broad range of methodological variation
Erratum in
-
Correction: Robustness of cancer microbiome signals over a broad range of methodological variation.Oncogene. 2024 May;43(20):1579. doi: 10.1038/s41388-024-03018-z. Oncogene. 2024. PMID: 38580705 Free PMC article. No abstract available.
Abstract
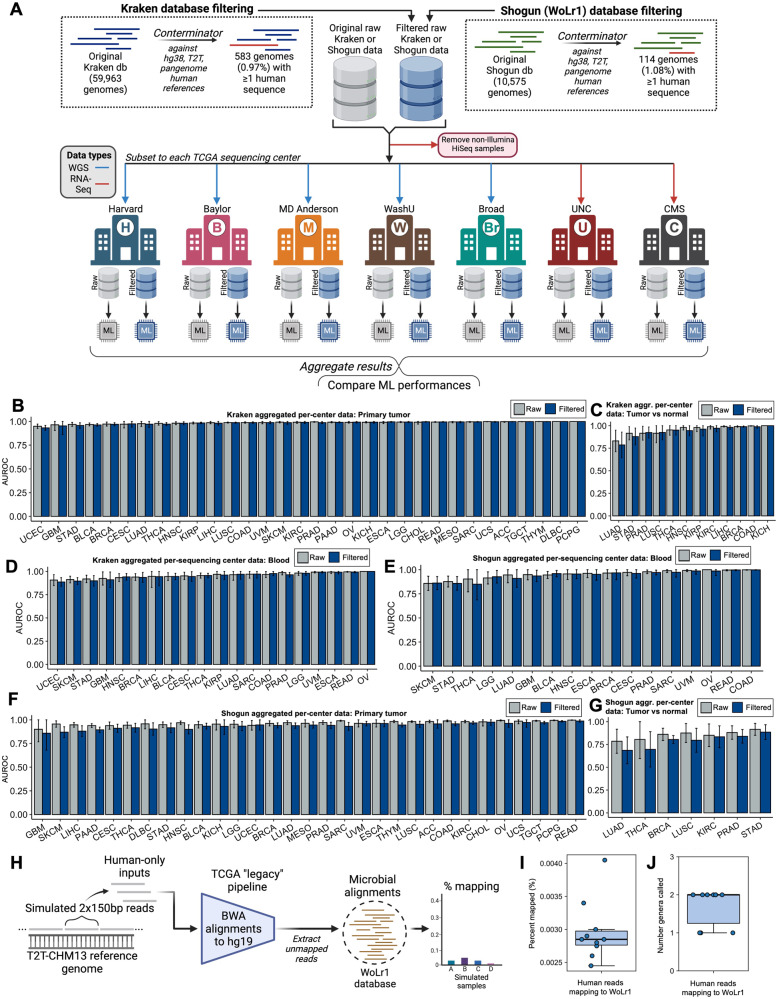
In 2020, we identified cancer-specific microbial signals in The Cancer Genome Atlas (TCGA) [1]. Multiple peer-reviewed papers independently verified or extended our findings [2-12]. Given this impact, we carefully considered concerns by Gihawi et al. [13] that batch correction and database contamination with host sequences artificially created the appearance of cancer type-specific microbiomes. (1) We tested batch correction by comparing raw and Voom-SNM-corrected data per-batch, finding predictive equivalence and significantly similar features. We found consistent results with a modern microbiome-specific method (ConQuR [14]), and when restricting to taxa found in an independent, highly-decontaminated cohort. (2) Using Conterminator [15], we found low levels of human contamination in our original databases (~1% of genomes). We demonstrated that the increased detection of human reads in Gihawi et al. [13] was due to using a newer human genome reference. (3) We developed Exhaustive, a method twice as sensitive as Conterminator, to clean RefSeq. We comprehensively host-deplete TCGA with many human (pan)genome references. We repeated all analyses with this and the Gihawi et al. [13] pipeline, and found cancer type-specific microbiomes. These extensive re-analyses and updated methods validate our original conclusion that cancer type-specific microbial signatures exist in TCGA, and show they are robust to methodology.
© 2024. The Author(s).
Conflict of interest statement
GDS-P and RK are inventors on a US patent application (PCT/US2019/059647) submitted by The Regents of the University of California and licensed by Micronoma; that application covers methods of diagnosing and treating cancer using multi-domain microbial biomarkers in blood and cancer tissues. GDS-P, RK, and SM-M are founders of and report stock interest in Micronoma. SF and SW are employees of Micronoma. GDS-P has filed several additional US patent applications on cancer bacteriome and mycobiome diagnostics that are owned by The Regents of the University of California and have been licensed by Micronoma. SF, SW, and GDS-P also have filed US patent applications related to cancer microbiome diagnostics that are owned by Micronoma. Additionally, RK is a member of the scientific advisory board for GenCirq, holds an equity interest in GenCirq, and can receive reimbursements for expenses up to US $5,000 per year; he is also an SAB member for DayTwo and BiomeSense, is a consultant for Cybele, is a co-founder of Biota, and owns equity in Biota, Cybele and BiomeSense. D.M. reports being a consultant and owning stock interest in BiomeSense. EK is a founder of Clarity Genomics. Clarity Genomics did not provide funding for this study. No disclosures were reported by the other authors.
Figures