

Pathological Interplay between Inflammation and Mitochondria Aggravates Glutamate Toxicity
- PMID: 38396952
- PMCID: PMC10889519
- DOI: 10.3390/ijms25042276
Pathological Interplay between Inflammation and Mitochondria Aggravates Glutamate Toxicity
Abstract
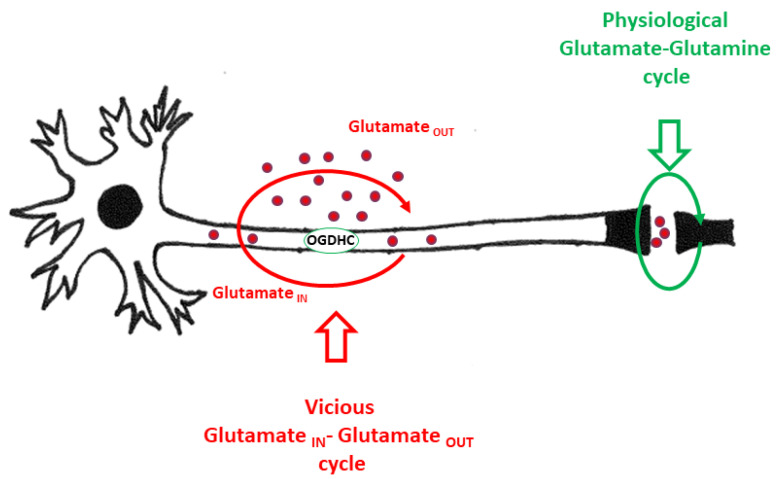
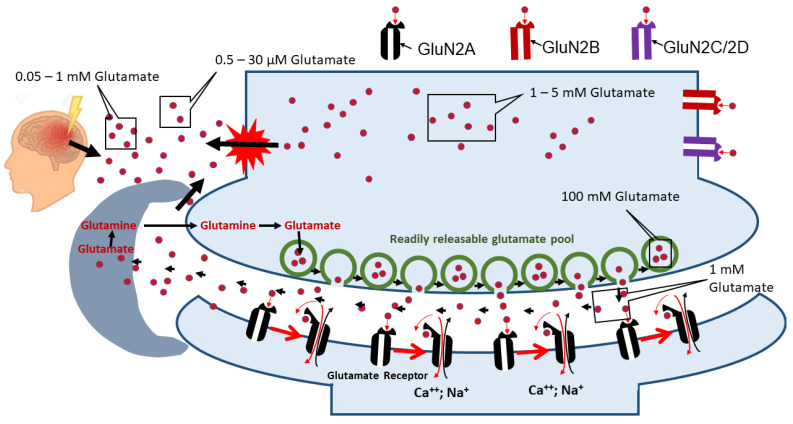
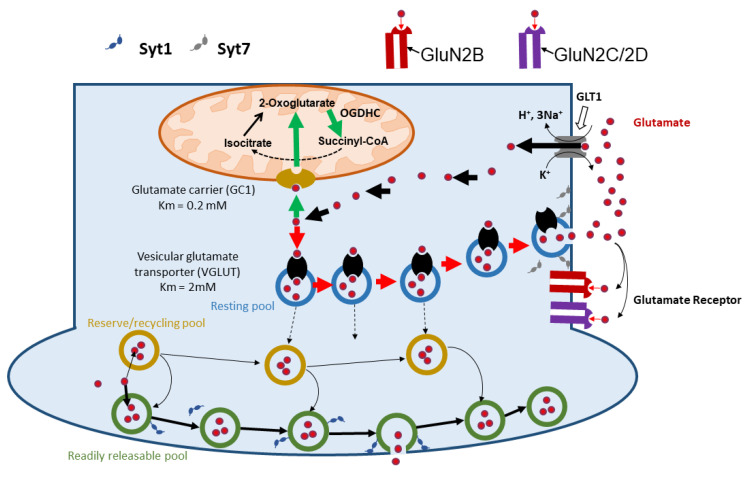
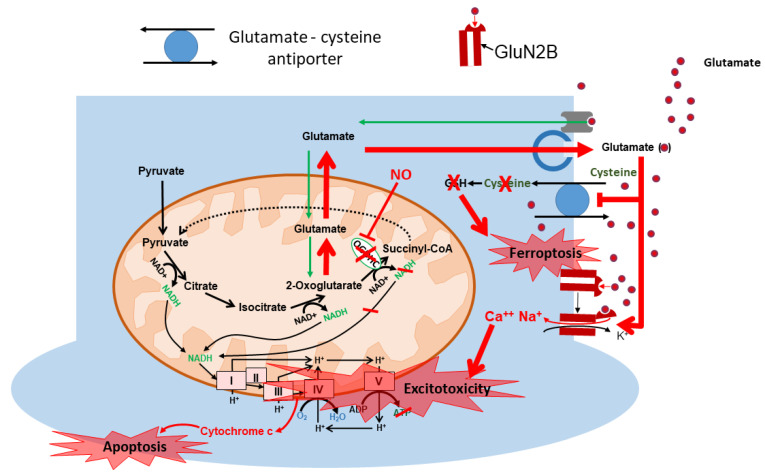
Mitochondrial dysfunction and glutamate toxicity are associated with neural disorders, including brain trauma. A review of the literature suggests that toxic and transmission actions of neuronal glutamate are spatially and functionally separated. The transmission pathway utilizes synaptic GluN2A receptors, rapidly released pool of glutamate, evoked release of glutamate mediated by Synaptotagmin 1 and the amount of extracellular glutamate regulated by astrocytes. The toxic pathway utilizes extrasynaptic GluN2B receptors and a cytoplasmic pool of glutamate, which results from the spontaneous release of glutamate mediated by Synaptotagmin 7 and the neuronal 2-oxoglutarate dehydrogenase complex (OGDHC), a tricarboxylic acid (TCA) cycle enzyme. Additionally, the inhibition of OGDHC observed upon neuro-inflammation is due to an excessive release of reactive oxygen/nitrogen species by immune cells. The loss of OGDHC inhibits uptake of glutamate by mitochondria, thus facilitating its extracellular accumulation and stimulating toxic glutamate pathway without affecting transmission. High levels of extracellular glutamate lead to dysregulation of intracellular redox homeostasis and cause ferroptosis, excitotoxicity, and mitochondrial dysfunction. The latter affects the transmission pathway demanding high-energy supply and leading to cell death. Mitochondria aggravate glutamate toxicity due to impairments in the TCA cycle and become a victim of glutamate toxicity, which disrupts oxidative phosphorylation. Thus, therapies targeting the TCA cycle in neurological disorders may be more efficient than attempting to preserve mitochondrial oxidative phosphorylation.
Keywords: TCA cycle; ferroptosis; glutamate; mitochondrial dysfunction; neuronal death.
Conflict of interest statement
The authors declare no conflicts of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
Figures
References
-
- Andrew R.D., Farkas E., Hartings J.A., Brennan K.C., Herreras O., Müller M., Kirov S.A., Ayata C., Ollen-Bittle N., Reiffurth C., et al. Questioning Glutamate Excitotoxicity in Acute Brain Damage: The Importance of Spreading Depolarization. Neurocrit. Care. 2022;37:11–30. doi: 10.1007/s12028-021-01429-4. - DOI - PMC - PubMed
-
- Mattson M.P. Stress: Physiology, Biochemistry, and Pathology Handbook of Stress Series. Volume 3. Elsevier; Amsterdam, The Netherlands: 2019. Excitotoxicity; pp. 125–134.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
